December 6, 2018
The paper I'll discuss in this post is this one: Extreme-ultraviolet refractive optics| Extreme-ultraviolet refractive optics (L. Drescher, O. Kornilov, T. Witting, G. Reitsma, N. Monserud, A. Rouzée, J. Mikosch, M. J. J. Vrakking & B. Schütte, Nature 564, 91–94 (2018) )
Lenses for focusing extreme UV light - just short of x-rays in energy - do not exist. One can imagine many applications for this capability, for instance in materials etching, welding and other processing, spectroscopic investigations of the structure of matter, cancer and other surgeries (should the focusing be refined enough - hard to see here since the lenses are gases) and regrettably, as no discovery is free of possible sinister use, weapons systems.
The introductory text tells the story better than I can:
Refraction of light is omnipresent in nature, where it forms the basis for the functionality of the human eye and the observation of a rainbow. It is exploited in many applications in the visible, infrared and ultraviolet spectral regions. For instance, refractive errors of the eye are corrected by glasses or contact lenses, and optical microscopes enable the magnification of small objects or structures. In the context of laser physics, refractive lenses are extensively used to focus or (de-)magnify laser beams. Dispersion and deflection of light by optical prisms is used to compress or stretch ultrashort laser pulses.
When Röntgen discovered X-rays in 1895, he attempted refraction experiments using prisms and lenses8. Because he observed no significant deflection of the X-rays, he concluded that refractive lenses were not suitable for focusing X-ray radiation. A century later, a compound refractive lens consisting of a lens array was nevertheless developed for the hard X-ray regime, assisted by the comparably low absorption in this spectral region. Compound refractive lenses are used to focus X-rays emitted from modern synchrotron9 and free-electron laser facilities10,11. They have been applied for hard X-ray microscopy12, for X-ray nanofocusing13 and for the investigation of crystal scattering14, as well as for coherent diffractive imaging of nanoscale samples15.
Refractive elements have so far been missing in the extreme-ultraviolet (XUV) range but are highly desirable. For instance, refractive lenses could be used to focus XUV pulses without changing the propagation direction, thereby providing considerable flexibility. The use of specially designed microscopic refractive lenses has been proposed16,17. However, the need to use very thin lenses with a sophisticated design, owing to the strong absorption of XUV radiation, makes practical implementation challenging.
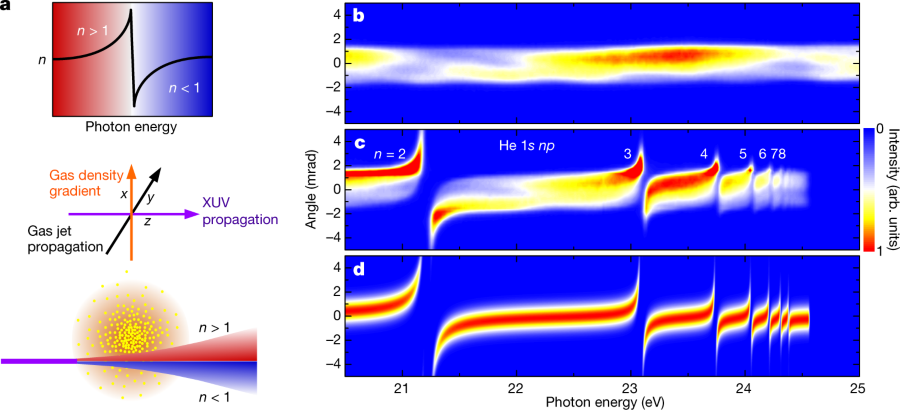
Here, we demonstrate that control over the refraction of XUV pulses can be achieved by using gases instead of solids. We exploit the fact that close to atomic resonances, the refractive index n has a dispersive lineshape, as depicted in the top part of Fig. 1a.
The authors utilize gas jets to focus the XUV beams using density gradients in the gas pulses. Since (in this case) the gases are monoatomic noble gases, they cannot be damaged by the energy that is contained in extreme UV radiation, as is the case with solid phase lenses.
If interested, take a look at the pictures.
Figure 1:
The caption:
a, Top, dispersive lineshape of the refractive index in the vicinity of an atomic resonance. Spectral components at photon energies below the resonance (n > 1) are indicated in red, components at energies above the resonance (n?<?1) in blue. Middle, experimental configuration, showing an XUV pulse (violet arrow) that crosses a gas jet (black arrow), which has a density gradient in the vertical direction (orange arrow), at right angles. Bottom, deflection of an XUV pulse propagating below the centre of the gas jet. b, Angle-resolved spectrum of a broadband HHG pulse measured in the absence of the gas jet. The angular divergence of the XUV beam in the vertical direction is reflected in the spatial distribution along the vertical axis. arb. units, arbitrary units. c, The same spectrum after propagation at a distance of 0.3 mm below the centre of a dense He gas jet (generated using a backing pressure of 10 bar) shows clear signatures of refraction. Spectral components with photon energies below the 1s np resonances of He are deflected upwards, whereas spectral components above these resonances are deflected downwards. The deflection angles are largest close to the 1s 2p resonance and decrease for higher resonances, owing to the decreasing oscillator strengths. Above the ionization potential of He (at 24.58 eV), the XUV radiation is strongly absorbed. Owing to ageing effects, the sensitivity of the detector was reduced in regions where the undisturbed HHG spectrum is recorded (as in b) compared with regions where the deflected XUV radiation is observed. This makes the deflected XUV radiation appear more intense. d, Simulation of the XUV refraction in an inhomogeneous He gas jet, taking into account 1s np resonances with n?=?2,?3,?…,?8. The simulation indicates that for a backing pressure of 10 bar, a gas jet with a peak density of 9?×?10^19 atoms cm?3 (corresponding to a pressure of 3.7 bar at 300 K) was achieved in the interaction zone.
Figure 2:
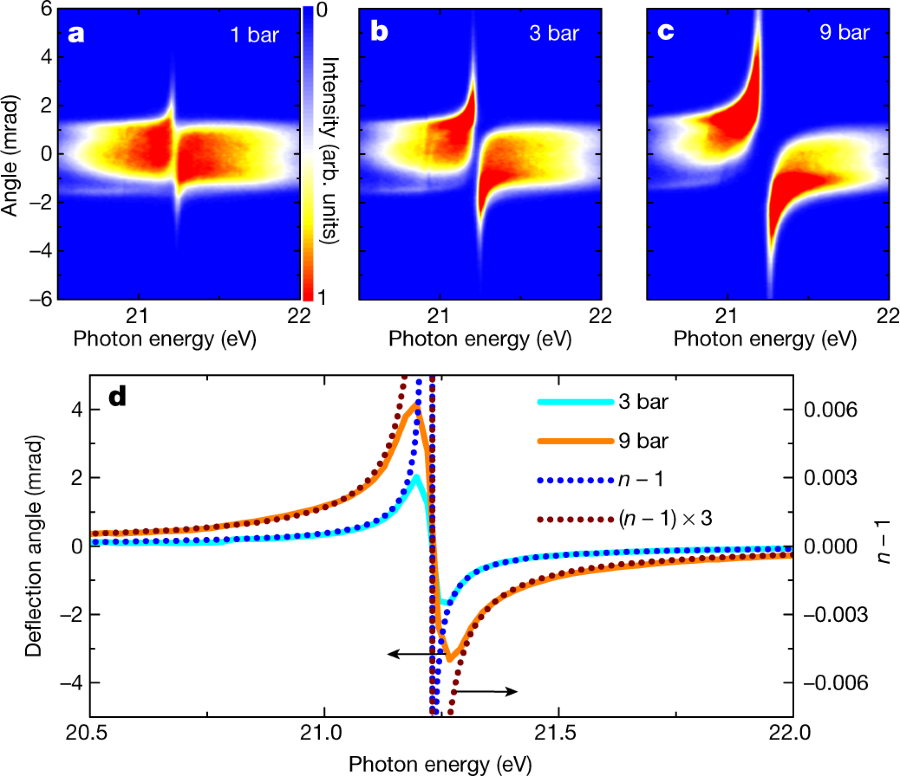
The caption:
a–c, Angle-resolved XUV spectra after propagation at a distance of 0.3 mm below the centre of a He gas jet, for backing pressures of 1 bar (a), 3 bar (b) and 9 bar (c). d, The average deflection angle as a function of the photon energy for backing pressures of 3 bar (corresponding to a peak pressure in the interaction zone of about 1 bar; cyan solid curve) and 9 bar (orange solid curve). Here the vertical scale on the left axis applies, as indicated by the upper arrow. For comparison, the calculated refractivity (that is, n ? 1) at standard temperature (273.15 K) and standard pressure (1 bar) is plotted on top of the deflection results (blue dotted curve). The vertical scale on the right axis applies, as indicated by the lower arrow. Note that the calculated refractivity is proportional to the pressure. The brown dotted curve shows the calculated refractivity multiplied by a factor of 3.
Image 3 (focused XUV radiation):
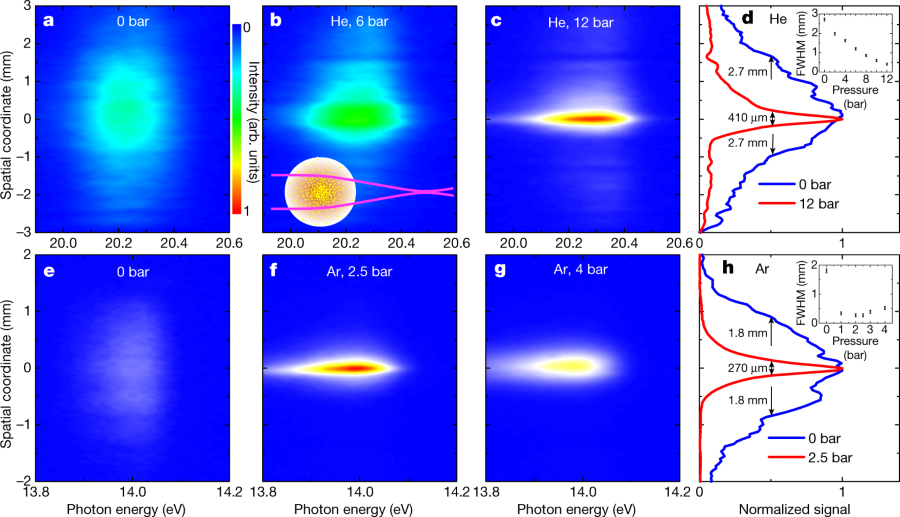
a, Spatially resolved spectrum of unfocused XUV radiation at 20.2 eV (corresponding to the 13th harmonic). b, c, The divergence of this harmonic is altered after propagation through a He gas jet (see inset of d), as shown for backing pressures of 6 bar (b) and 12 bar (c). d, Comparison of the vertical beam profiles using backing pressures of 0 bar (blue curve) and 12 bar (red curve). The inset shows the pressure-dependent spot size, where the error bars reflect the uncertainties in determining the spot sizes. e, Spatially resolved spectrum of radiation at 14.0 eV (corresponding to the ninth harmonic), which is close to the 3d and 5s resonances of Ar. f, Focusing of this harmonic is achieved by an Ar gas jet at a backing pressure of 2.5 bar. g, When further increasing the backing pressure to 4 bar, an increasing beam size is observed, because the Ar lens focuses the XUV beam between the gas jet and the detector. h, The vertical beam profiles for Ar backing pressures of 0 bar (blue curve) and 2.5 bar (red curve). The inset shows the pressure-dependent spot size, where the error bars reflect the uncertainties in determining the spot sizes.
Fig. 4: Simulation of the XUV focus:
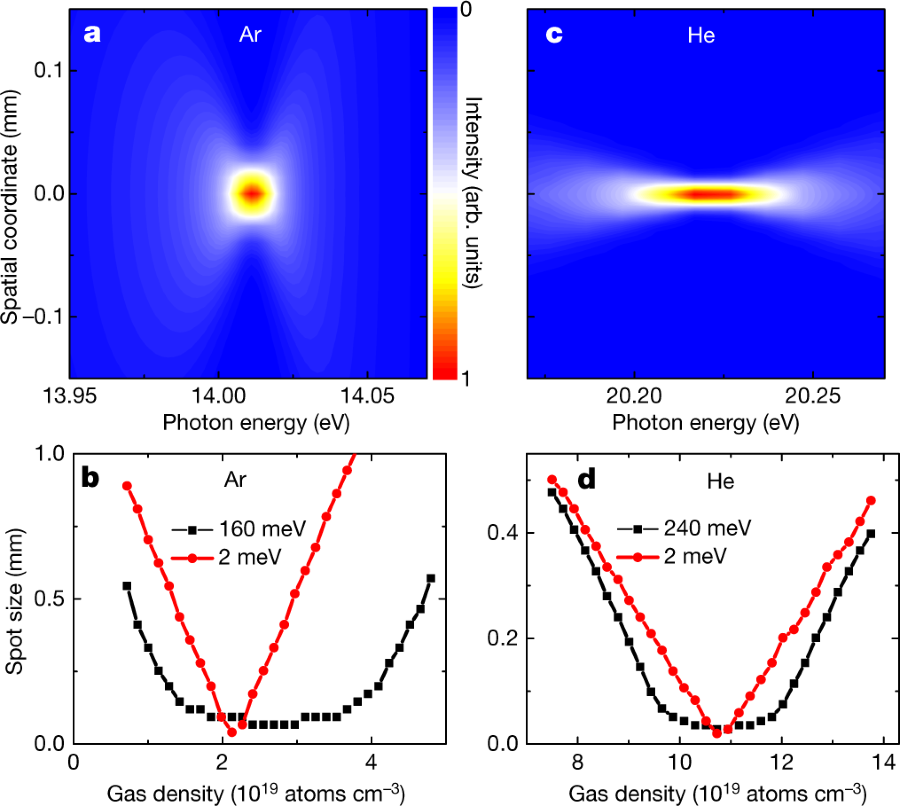
The caption:
a, Simulated focus in the vertical direction as a function of the photon energy following propagation of an XUV pulse at 14.0 eV (1.9 mm FWHM diameter) through an Ar gas jet with a peak density of 2.2?×?1019 atoms cm?3 (corresponding to a pressure of 0.9 bar at 300 K). Because of chromatic aberration, the XUV spot size depends on the photon energy. b, Spot size as a function of the Ar gas density for XUV pulses with a bandwidth of 160 meV (black curve) and 2 meV (red curve), showing minimal spot sizes of 74 ?m and 40 ?m, respectively. c, The chromatic aberration is reduced for photon energies that are further away from the resonance. This is shown for the example of an XUV pulse at 20.2 eV (2.4 mm FWHM diameter) that propagates through a He gas jet with a peak density of 1.1?×?1020 atoms cm?3 (corresponding to a pressure of 4.3 bar at 300 K). d, Spot size as a function of He gas density for XUV pulses with a bandwidth of 240 meV (black curve) and 2 meV (red curve), which exhibit minimal spot sizes of 28 ?m and 20 ?m.
The authors conclude:
In conclusion, we have presented a method to deflect and focus XUV pulses by using the inhomogeneity of a gas jet placed in the way of an XUV beam. Our results enable the transfer of concepts based on refractive optics that are widely used in other spectral regions to the XUV regime, including microscopy, nanofocusing and the compression of ultrashort pulses. XUV gas-based lenses have several advantages, including their high transmission, deformability and tunability (by varying the gas composition, the gas pressure and the gas jet geometry)...
...Refractive XUV gas-phase lenses can be designed for photon energies between 10 eV and 24 eV by carefully selecting appropriate atoms or molecules for different photon energies. In the future, this range might be extended to higher photon energies by developing lenses that exploit refraction in an inhomogeneous plasma consisting of highly charged ions and electrons.
Esoteric, but interesting.
Have a pleasant day tomorrow.
December 4, 2018
The paper I'll discuss in this post is this one: Methicillin-resistant Staphylococcus aureus alters cell wall glycosylation to evade immunity. (Peschel, Stehel et al Nature 563, pages705–709 (2018) )
A kind of mental block I've had over the years involves my understanding of the complex chemistry and biochemistry of sugars.
I've always found it hard, and tend to squirrel around it when it comes before me.
It's why I always love to read the introductory text of Gabius' The Sugar Code which I'm sure I've referenced before in this space.
To wit:
Teaching the biochemistry of carbohydrates is not simply an exercise in terminology. It has much more to offer than commonly touched upon in basic courses, if we deliberately pay attention to the far-reaching potential of sugars beyond energy metabolism and cell wall stability. In fact, then there is no reason why complex carbohydrates should shy at competition with nucleic acids and proteins for the top spot in high-density biocoding. On the contrary, sugars have ideal properties for this purpose, as will be concluded at the end of this chapter. In this sense, an obvious explanation why research in glycosciences (structural and functional glycomics and lectinomics) has lagged behind the fields of genomics and proteomics, also in the public eye, is 'that glycoconjugates are much more complex, variegated, and diffiailt to study than proteins and nucleic acids' [1]. What is a boon for decorating cell surfaces with a maximum number of molecular messages at the same time has been and still is a demanding challenge for analytical and synthetic chemistry…
H.J. Gabius ed. The Sugar Code, Wiley, 2009 pg 1.
That about sums it up. People don't think about sugars too much, because it's hard to do so...
(It's a great book by the way. I have to reference it all the time.)
One of the great - among many - challenges for future generations is antibiotic resistance. The extension of life span around the world is to a large measure the existence of antibiotics. My grandmother died at the age of 39 from a simple infection that could be cured today with a trip to the doctor, a prescription, and maybe 20 or 30 pills.
In order to address this rising crisis, it is useful to know
how bacterial evolve to develop resistance to drugs, and, for that matter, natural antibodies.
This is why I found the paper cited above quite interesting.
From the introduction:
Novel prevention and treatment strategies against major antibiotic-resistant pathogens such as MRSA are urgently needed but are not within reach because some of the most critical virulence strategies of these pathogens are not understood8. The pathogenic potential of prominent healthcare-associated (HA)-MRSA and recently emerged livestock-associated (LA)-MRSA strains is thought to rely on particularly effective immune evasion strategies, whereas community-associated (CA)-MRSA strains often produce more aggressive toxins1,2. Most humans have high overall levels of antibodies against S. aureus as a consequence of preceding infections, but antibody titres differ strongly for specific antigens and are often not protective in immunocompromised patients, for reasons that are not clear3. A large percentage of human antibodies against S. aureus is directed against WTA5,9,10, which is largely invariant. However, some S. aureus lineages produce altered WTA, which modulates, for instance, phage susceptibility 7,11...
WTA is "Wall teichoic acid."
Teichoic acid, like nucleic "acid" is actually a copolymer.of a doubly phosphorylated acetylated aminosugar, galactosamine, and glycerol (or ribotol, an alcohol formed by reducing ribose.)
Here's a structure from Wikipedia:

The authors identified a protein called "TarP" that has close homology (27%) to a protein found in "normal" (not resistant) staphylococcus TarS. When they reinserted TarS into resistant strains, they found that they could restore susceptibility to antibiotics, as well as manipulate susceptibility to certain viruses.
I don't have a lot of time tonight, but here's some pictures and captions from the paper:
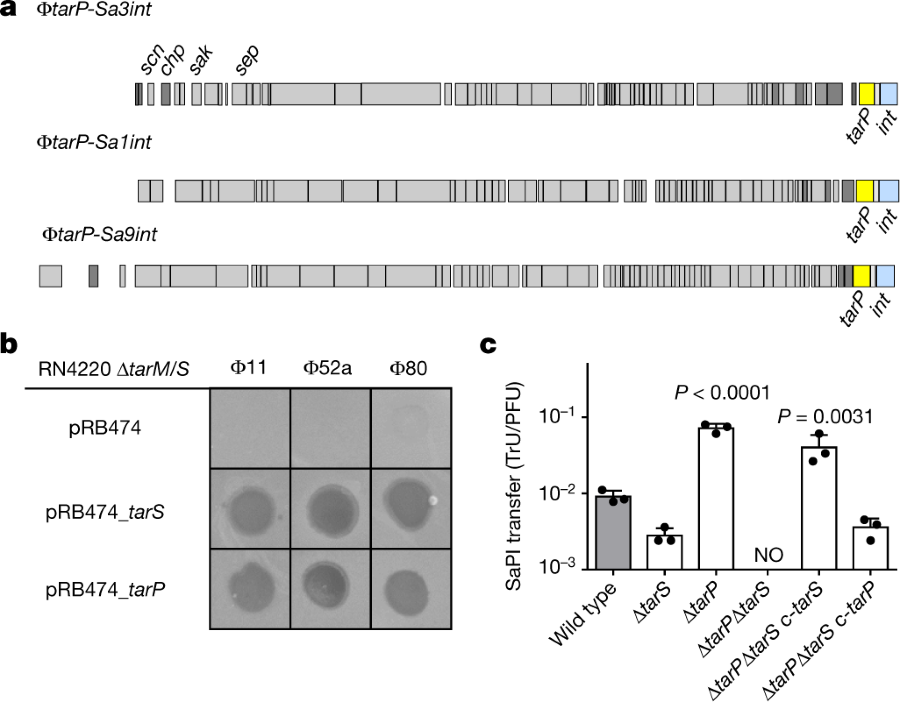
The caption:
a, TarP is encoded next to different integrase types (int gene) in prophages ?tarP-Sa3int (with immune evasion cluster scn, chp, sak, sep), found in HA-MRSA, and ?tarP-Sa1int and ?tarP-Sa9int, identified in LA-MRSA. TarP variants in ?tarP-Sa1int and ?tarP-Sa9int differed from TarP in ?tarP-Sa3int in one amino acid each (I8M and D296N, respectively). Both residues are distant from the catalytic centre. b, Complementation of S. aureus RN4420 ?tarM/S with either tarS or tarP restores susceptibility to infection by WTA GlcNAc-binding siphophages, as indicated by plaque formation on bacterial lawns. Data shown are representative of three independent experiments. c, tarP expression reduces siphophage ?11-mediated transfer of SaPIbov in N315. Values indicate the ratio of transduction units (TrU) to plaque-forming units (PFU) given as mean?±?s.d. of three independent experiments. Statistical significances when compared to wild type were calculated by one-way ANOVA with Dunnett’s post-test (two-sided) and significant P values (P???0.05) are indicated. NO (none obtained) indicates no obtained transductants.
Some protein structures, teichoic acid structures, and some NMR's:
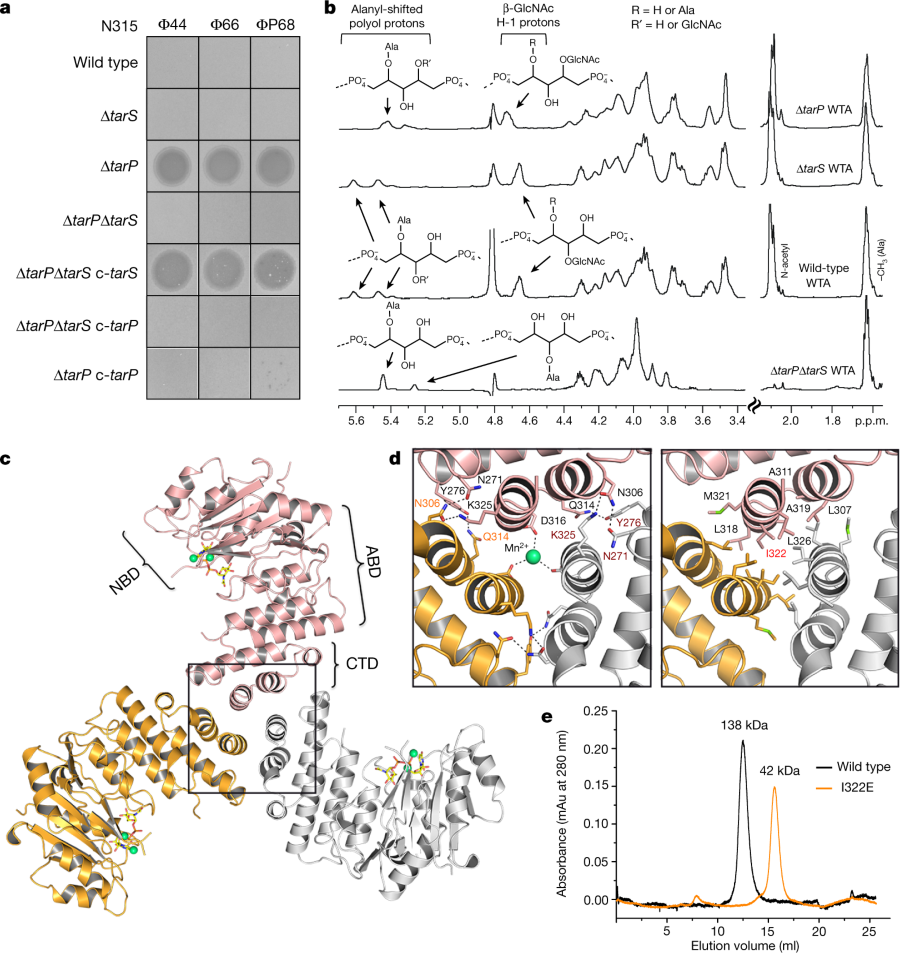
The caption:
a, Expression of tarP renders N315 resistant to podophages. Representative data from three independent experiments are shown. b, 1H NMR spectra reveal different ribitol hydroxyl glycosylation of N315 WTA by TarS (C4) or TarP (C3). The RboP units with attached GlcNAc are depicted above the corresponding proton resonances. Representative data from three experiments are shown. In-depth description of the structural motifs identified in the spectra is given in the Supplementary Information. c, Crystal structure of TarP homotrimer (pink, orange, grey) bound to UDP-GlcNAc (yellow) and two Mn2+ ions (lime green). The nucleotide-binding domain (NBD), acceptor-binding domain (ABD), and C-terminal trimerization domain (CTD) of the pink monomer are labelled. d, Views into the trimer interface (boxed in c). Left, polar interactions. Hydrogen bonds and salt bridges are shown as black dashed lines. The Mn2+ is 2.1 Å from each Asp316 carboxylate. Right, hydrophobic interactions, with the mutated residue Ile322 highlighted in red. e, Size-exclusion chromatography elution profiles. Based on calibration of the column, the TarP wild-type and I322E mutant proteins have estimated molecular weights of 138 kDa (n?=?8) and 42 kDa (n?=?3), respectively, in agreement with the calculated molecular weights of 120 kDa for a TarP trimer and 40 kDa for monomeric TarP.
A cartoon of interactions leading to the modified cell walss:
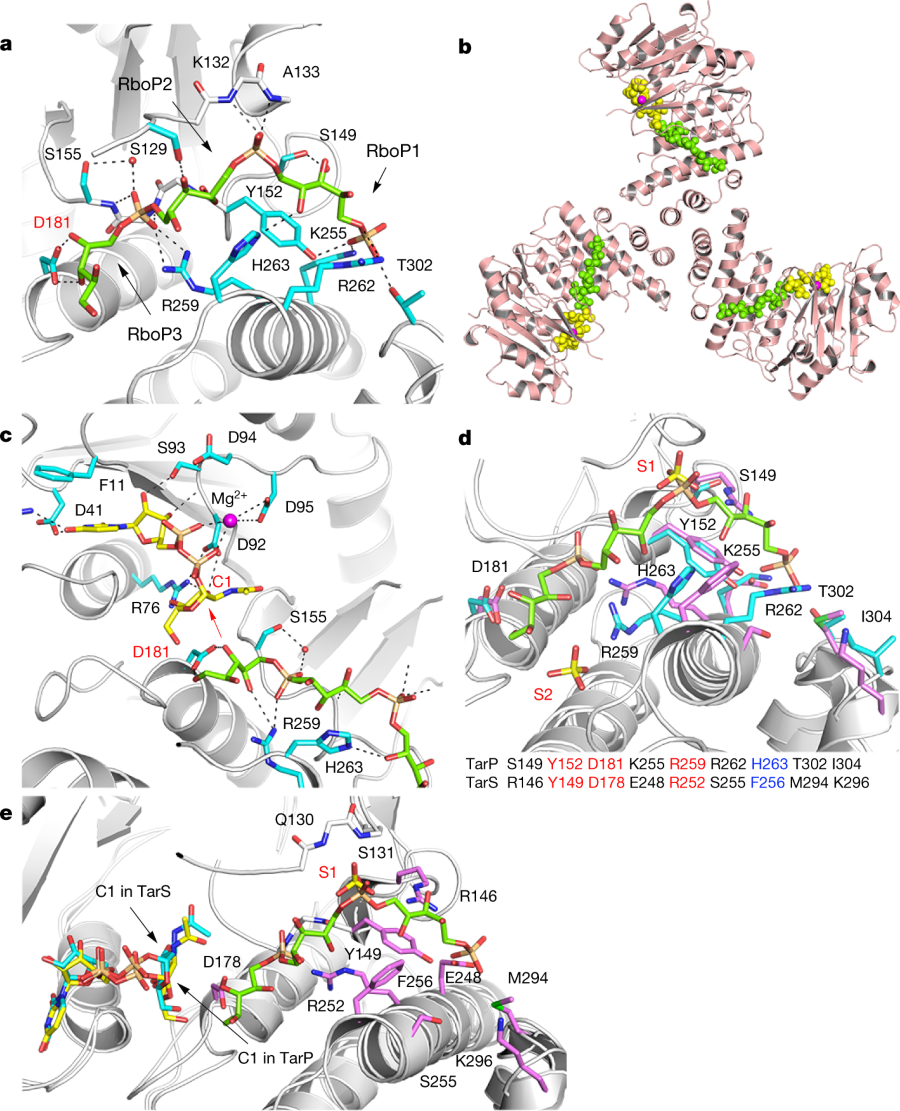
a, 3RboP binding site in the TarP–3RboP complex, with key amino acids shown (cyan). Asp181 is highlighted in red. The ribitol of 3RboP is coloured green and D-ribitol-5-phosphate units 1, 2 and 3 (RboP1, RboP2, and RboP3) are labelled. Hydrogen bonds and salt bridges are shown as black dashed lines. b, Ternary complex of TarP with UDP-GlcNAc and 3RboP. UDP-GlcNAc, Mg2+ and 3RboP are shown as full-atom models coloured yellow, magenta, and green, respectively. c, View into the active site of TarP. C1 of UDP-GlcNAc and Asp181 are highlighted with red labels. The arrow indicates how the C3-hydroxyl in RboP3 could nucleophilically attack GlcNAc C1. d, Comparison of the polyRboP-binding site of TarP with the corresponding region in TarS. Residues of TarP and 3RboP are coloured as in a. TarS residues are coloured violet and the two sulfates are labelled S1 and S2. Only residues of TarP are labelled, for clarity. Key TarP and TarS residues lining the polyRboP-binding site are shown at the bottom, with three identical (red) and one conserved amino acids (blue). e, Superposition of UDP-GlcNAc-bound TarS with the ternary TarP–UDP-GlcNAc–3RboP complex. UDP-GlcNAc and 3RboP bound to TarP are coloured as in b, whereas UDP-GlcNAc bound to TarS is coloured in cyan. Only the TarS residues are shown (coloured as in d), for clarity. The arrows indicate the C1 positions of UDP-GlcNAc in TarP and TarS.
Some immunology effects:
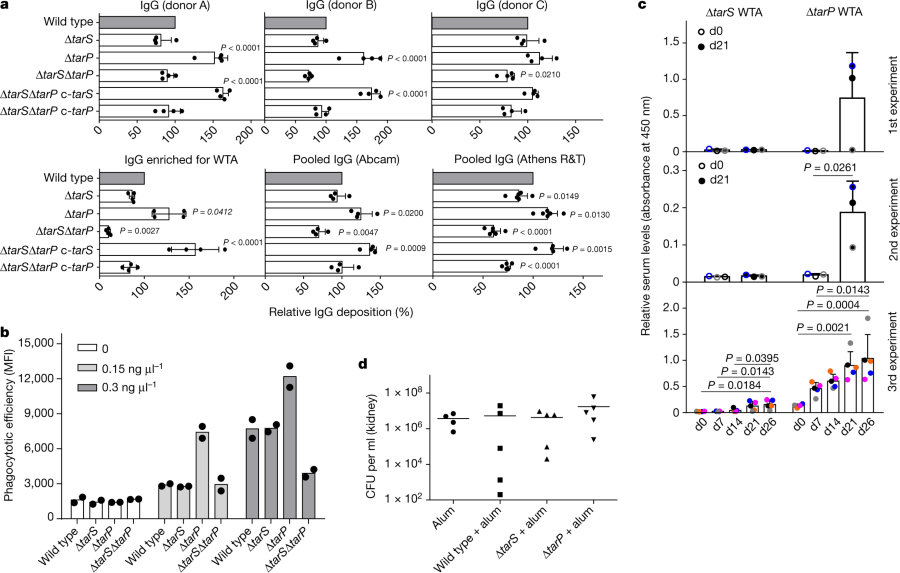
The caption:
a, TarP expression reduces deposition of IgG from human serum on N315 cells. The protein A gene spa was deleted in all strains. Top, human IgG isolated from three individual healthy donors (A, B, and C; n?=?4); bottom, left, IgG from human serum enriched for RN4220 WTA binding (n?=?4); middle and right, pooled human IgG from different suppliers (Abcam, n?=?4; Athens R&T, n?=?6). Results were normalized against wild type and shown as means with s.d. of n experiments. P values for comparison with wild type were calculated by one-way ANOVA with Dunnett’s post-test (two-sided), and P???0.05 was considered significant. Significant P values are displayed. b, TarP reduces neutrophil phagocytosis of N315 strains lacking protein A, opsonized with indicated concentrations of IgG enriched for WTA binding. Values are depicted as mean fluorescence intensity (MFI). Means of two dependent replicates of a representative experiment are shown. The other two representative experiments can be found in Extended Data Fig. 1g. c, TarP abrogates IgG response of mice towards WTA. For each experiment, WTA from N315 ?tarP or ?tarS was isolated independently. At least three mice per group were vaccinated and analysed for specific IgG at indicated time points after vaccination. Results are depicted as mean absorbance with s.d. Individual mice are indicated by colour. Increase in IgG levels was assessed by one-way ANOVA with Tukey’s post-test (two-sided). Significant differences (P???0.05) are indicated with corresponding P values. d, Vaccination with WTA does not protect mice against tarP-expressing N315, as shown for bacterial loads in kidney upon intravenous infection. No significant differences between groups of either five vaccinated mice or four mice for the alum control group (means indicated), calculated by one-way ANOVA, were observed.
Some concluding remarks:
Protection against S. aureus infections is urgently needed, in particular for hospitalized and immunocompromised patients2,4. Antibodies can in principle protect against S. aureus, but their titres and specificities vary largely among humans and they are often not protective in immunocompromised patients3, probably in particular against S. aureus clones that mask dominant epitopes, for instance using TarP. Unfortunately, all previous human vaccination attempts with protein or glycopolymer antigens have failed, for reasons that are unclear24. Our study identifies a new strategy used by pandemic MRSA clones to subvert antibody-mediated immunity, which should be considered in future vaccination approaches. S. aureus WTA with GlcNAc at RboP C3 has been reported as a type-336 antigen, but was not further explored25. We found that tarP is present in type-336 S. aureus (Extended Data Fig. 1f). However, TarP-modified WTA is a very poor antigen and vaccines directed against GlcNAc at WTA RboP C3 or C4 may fail against many of the pandemic MRSA clones. The structural characterization of TarP will instruct the development of specific TarP inhibitors that could become important in combination with anti-WTA vaccines or antibiotic therapies...
...TarP is a new and probably crucial component of the S. aureus virulence factor arsenal26,27, highlighting the important roles of adaptive immunity and its evasion in S. aureus infections.
A note, which may or may not be all that easy to comprehend, from the front lines in confronting the growing antibiotic crisis with molecular biology.
Have a pleasant day tomorrow.
December 2, 2018
The paper I'll discuss in this post is this one: Pentavalent Curium, Berkelium, and Californium in Nitrate Complexes: Extending Actinide Chemistry and Oxidation States (Attila Kovács,*,† Phuong D. Dau,‡ Joaquim Marçalo,§ and John K. Gibson*, Inorg. Chem., 2018, 57 (15), pp 9453–9467.
The shape of the periodic table is actually a quantum mechanical effect. Every electron in an atom must have a unique set quantum configuration numbers, defined by it's primary shell number (which are represented by the rows in the periodic table), and then its suborbital, usually designated for historical reasons as s, p, d, and f, which are represented by its position in a column relative to the "steps" that appear in the table's shape.
In the periodic table both the lanthanides and actinides, the "f elements," appear in the boxes below the main group elements, and the discovery that the actinides, in particular, should go there was the recognition by Glenn Seaborg that they were "f elements" and not, as previously thought as "d elements" that begin with Scandium (Sc) and end with the synthetic element Copernicium (Cn), the "d block elements. The "d block" is actually broken by elements La-Lu (Lanthanum to Lutetium) and Ac-Lw (Actinium to Lawrencium). In fact the "f elements" should represent another "step" in the periodic table, but printing it in this way is logistically difficult since it would be difficult to print on standard paper without making the print too small to read, so they're put in boxes below the "main group" elements.
The heaviest element that has been isolated in a relatively pure form in quantities that are visible is element 99, Einsteinium. It seems theoretically possible to isolate, perhaps, albeit at enormous expense, a visible, if transitory, sample of fermium, element 100, since it is the last element formed by sequential beta decay, but I don't believe it has ever been done, nor will it ever be done. Generally fermium and all of the elements beyond are synthesized on an atom by atom scale in accelerators and are basically known from their decay products and the high energy radiation they produce.
The lanthanide elements, with a few important exceptions generally exhibit the +3 oxidation state, although a few elements like cerium (+4) and europium and samarium (+2) have other oxidation states, but they are all mostly characterized by +3 oxidation state, making their separations from one another somewhat difficult, meaning that their industrial chemistry, important in many modern devices, is at best environmentally suspect at best, environmentally odious at worst.
The chemistry of the lower actinide elements, including those that naturally occur if far richer. In fact thorium is almost always found in the +4 state, protactinium in the +5 state, and uranium in either the +4 or +6 state in the natural environment. For a long time, before Seaborg's discovery, these elements were thought to be "d elements" and in fact, thorium has chemistry much closer to zirconium and hafnium than say, curium, protactinium is more "tantalum like" than curium like, and uranium has many similarities to tungsten. (The presence of billion ton quantities of uranium in oceans only became possible on earth after oxygen appeared in the atmosphere, resulting in the somewhat more soluble +6 uranium oxidation state being formed by oxidation as opposed to the very insoluble +4 state. Uranium, and plutonium, but not generally neptunium, have well characterized +3 states, but thorium, protactinium, do not. (Uranium, neptunium, and plutonium all exhibit volatile hexafluorides (+6) albeit of decreasing stability in sequence; a fact of industrial importance; in the oceans and in certain fresh water supplies, uranium VI is present as the dioxo ion.)
In nuclear technology, the existence of multiple oxidation states among actinide elements greatly simplifies their separations from one another (but not necessarily from fission products), at least in the case where there are only trivial amounts of the transamericium elements, curium and berkelium and californium, all of which can be isolated in gram quantities, and in a the case of curium, kg quantities.
I personally always assumed that except for some exotic chemistry involving +2 states for curium at least, that curium, berkelium and californium most commonly exhibited +3 chemistry and that no higher states existed.
I was wrong.
From the paper cited above:
The range of accessible oxidation states (OSs) of an element is fundamental to its chemistry. In particular, high OSs provide an assessment of the propensity, and ultimately the ability, of valence electrons to become engaged in chemical bonding. Until recently, the highest known OS in the entire Periodic Table was VIII, such as in the stable and volatile molecules RuO4 and OsO4. The OS IX was finally realized in the gas-phase complex IrO4+,(1) but neither this moiety nor this highest OS has been isolated in the condensed phase.(1,2) The appearance of distinctive and otherwise inaccessible chemistry in gas-phase species, such as IrIX in IrO4+, is generally attributed to isolation of a moiety that would otherwise be highly reactive with neighbor species in condensed phases.(3) For example, gas-phase PaO22+, which comprises formally PaVI, has been synthesized, but it activates even dihydrogen to yield atomic H and PaO(OH)22+ in which the stable discrete PaV OS state is recovered.(4) In view of its gas-phase reactivity, there is scant chance of isolating PaO22+ in the condensed phase. Another example of a distinctively high OS accessible (so far) only in the gas phase is PrV in PrO2+ and NPrO,(5,6) this being the only known pentavalent lanthanide.
The early actinides yield ultimate OSs, from AcIII to NpVII, that correspond to engagement of all valence electrons in chemical bonding to yield an empty 5f0 valence electron shell.(7) After Np, the highest accessible actinide OSs, from PuVII to lower OSs beyond Pu, have one or more chemically unengaged valence 5f electron(s), as the nuclear charge increases and energies of the 5f orbitals decrease. The transition from chemical participation of all 5f valence electrons in ubiquitous UVI, to participation of only two valence electrons in prevalent NoII,(8) distinguishes the actinides from the lanthanides for which the relatively low energy of the valence 4f orbitals results in only a few OSs above trivalent.(9) The gas-phase molecular ions BkO2+ and CfO2+ were recently synthesized and their OSs computed as BkV and CfV, which was an advancement beyond oxidation state IV for these elements and extended the distinctive actinyl(V) dioxo moieties into the second half of the actinide series.(10) It is notable that the computed oxidation state in ground-state CmO2+ is not CmV but rather CmIII, which reflects the limited stabilities of OSs above III for the actinides after Am.(10)
A primary goal of the work reported here is to assess stabilities of OSs, particularly the pentavalent OS, of the actinides Cm, Bk, and Cf. These elements represent the transition from the early actinides that exhibit higher OS, including AmVI and possibly also AmVII,(11) to the latest actinides, Es through Lr, that have been definitively identified only in the AnII and/or AnIII OS. The meagre realm of OSs for the late actinides may not be entirely due to intrinsic chemistry because synthetic efforts for these elements have been very limited due to scarcity and short half-lives of available synthetic isotopes. Cm, Bk, and Cf are the heaviest actinides available as isotopes that are both sufficiently abundant (>10 ?g) and long-lived (>100 days) for application of some conventional experimental approaches with relatively moderate procedural modifications.
The higher oxidation states were synthesized in the gas phase by the use of electrospray ionization (ESI) and detected in the mass spectrometer in which the ESI was performed.
The results of the spectra were verified by quantum mechanical computations using
AIMAll Software
Some cool pictures from the paper:

The caption:
Scheme 1. Generic D2h, C2v, and C2 Symmetry Structures for AnO2(NO3)2–
Apparently this technique has also been applied to lanthanides, motivating this work:

Figure 1. CID mass spectra acquired at a nominal instrumental voltage of 0.5 V for (a) Ce(NO3)4–, (b) Pr(NO3)4–, (c) Nd(NO3)4–, and (d) Tb(NO3)4–. Elimination of NO2 is indicated by arrows. Sequential CID elimination of two NO2 is observed only for Pr(NO3)4– to yield PrO2(NO3)2–.
Mass spectra from the actinides:

The caption:
Figure 2. CID mass spectra acquired at a nominal instrumental voltage of 0.5 V for (a) Pu(NO3)4–, (b) Am(NO3)4–, (c) Cm(NO3)4–, (d) Bk(NO3)4– (with 7% isobaric Cf(NO3)4– from beta-decay of 249Bk), and (e) Cf(NO3)4–. Elimination of NO2 is indicated by arrows. Sequential CID elimination of two NO2 molecules from An(NO3)4– to yield AnO2(NO3)2– is observed in all five cases.
Some calculated structures:

The caption:
Figure 3. Structures of CfIVO2(NO3)2– (top) and CfIIIO2(NO3)2– (bottom) in two perspectives and selected distances in angstrom from CASPT2/DZ calculations.
Results of density functional theory calculations for a curium oxonitride complex:

The caption:
Figure 4. Electron density map of CmO2(NO3)2– from DFT calculations. Charge concentration is indicated by yellow, while charge depletion is indicated by blue.
Molecular orbitals for the plutonium complex in this class:

The caption:
Figure 5. Characteristic molecular orbitals of PuO2(NO3)2– from CASPT2 calculations
The same thing for Berkelium:

The caption:
Figure 6. Characteristic molecular orbitals of BkO2(NO3)2– from CASPT2 calculations.

A text excerpt:
Of the various theoretical approaches, only the AIM model can characterize quantitatively the space between the bonding atoms. Therefore, we performed a topological analysis of the electron density distribution of the AnVO2(NO3)2– complexes in order to see how the density properties of the An–O bonds vary along the 5f row. We were particularly interested in the parameters of An–nitrate interactions, as they may provide a clue on the increasing bend along the actinide row. A graphical representation of the bonding paths, bond and ring critical points of AmVO2(NO3)2– is shown in Figure 7.
Figure 7:
The caption:
Figure 7. Bonding paths (black), bond (green), and ring (small red) critical points of AmVO2(NO3)2–.
Ionization energies:

The caption:
Figure 10. Actinide ionization energies in eV(79) (using corrected value for IE[U3+] as discussed above): (a) fourth IE; (b) fifth IE; (c) sum of fourth and fifth IEs. Dotted lines are approximate upper stability boundaries for (a) AnIV relative to AnIII; (b) AnV relative to AnIV; (c) AnV relative to AnIII.
Some remarks from the conclusion:
Comparison of experimental results for lanthanide and actinide oxide nitrate anion complexes suggested the AnV oxidation state as coordinated actinyl(V) moieties embedded in AnO2(NO3)2– for An = Pu, Am, Cm, Bk, and Cf, this being the first Cm(V) complex. The stability of oxidation state V in these AnO2(NO3)2– complexes has been confirmed by quantum chemical calculations. The relative stability of this oxidation state is particularly notable for Cf and Bk complexes, and therefore the AnIVO2(NO3)2– and AnIIIO2(NO3)2– forms have been explored and their lower stabilities with respect to CfV and BkV have been supported by both CASPT2 and DFT calculations. Whereas pentavalent Cf was expected to be stable due to a half-filled 5f7 configuration, the computations show that this configuration for CfVO2(NO3)2– is not octet with all seven 5f electrons spin-unpaired, but rather sextet with two of the 5f electrons spin-paired in a 5f1+ orbital.
The AnO2(NO3)2– complexes show interesting bonding features. While in the actinyl moiety the ionic character of bonding increases from Pu to Cf (in agreement with experience on several other actinide systems), in the An–NO3– interaction an opposite trend has been observed here. The increasing ionicity in the AnO2 moiety results in charge depletion around An making it more suitable as acceptor for charge transfer from the nitrate oxygens. The increasing covalent character from Pu to Bk ? Cf may be an important factor for the trend observed in the molecular geometries, i.e., a gradual bend of the NO3– ligands (described by the N–An–N angle) around An...
I'm well aware that this may all seem very "out there," and perhaps, in some quarters, generate remarks along the lines of "I couldn't care less."
I assure you though, whether you are inclined to believe it or not, or even if you despise the idea, that the chemistry of the actinides is critical, absolutely critical, to saving whatever is left to save of our rapidly deteriorating environment.
I wish you a rather pleasant Sunday.
December 1, 2018
From Nature: Osamu Shimomura (1928–2018)
Growing up during one of the darkest times in history, Osamu Shimomura devoted his long and fruitful career to understanding how creatures emit light. He discovered green fluorescent protein (GFP), with which — decades later — biomedical researchers began to monitor the workings of proteins in living tissue, and to confirm the insertion of genes. For that discovery, he shared the Nobel Prize in Chemistry in 2008 with neurobiologist Martin Chalfie and the late Roger Tsien, a chemist.
Shimomura, who died in Nagasaki, Japan, on 19 October, was the first to show that a protein could contain the light-emitting apparatus within its own peptide chain, rather than interacting with a separate light-emitting compound. The significance of this discovery was that the gene encoding GFP could, in principle, be copied (or ‘cloned’) and used as a tool in other organisms...
...Born on 27 August 1928 in the town of Fukuchiyama, at the height of Japanese expansionism, Shimomura was the son of an army captain whose frequent postings abroad disrupted his child’s school education. Shimomura’s grandmother instilled in him the samurai principles of honour and fortitude. In 1944, with the Pacific War turning against Japan, he and his fellow school students were mobilized to work in a munitions factory in Isahaya, about 25 kilometres from Nagasaki. On 9 August 1945, he was at work when a blinding flash, followed by a huge pressure wave, signalled the dropping of the atomic bomb on the nearby city. He walked home under a shower of black rain. He later wrote that his grandmother’s quick action in putting him straight in the bath might have saved him from the effects of the radiation.
Without a high-school diploma, he despaired of finding a college place. Eventually, Nagasaki Pharmacy College admitted him in 1948. On graduation, he worked for four years as an assistant in practical classes. He devised research projects in his own time, and his professor obtained permission for him to do a year of advanced study...
...The luciferin paper brought an invitation for Shimomura to join the bioluminescence lab of biologist Frank Johnson at Princeton University in New Jersey. Three weeks after marrying Akemi Okubo in August 1960, Shimomura sailed to the United States, his travel paid for by a Fulbright scholarship...
...He discovered almost at once that it was activated by calcium (later, aequorin became an essential reagent as a glowing marker of calcium release). Shimomura, his family and his research colleagues spent 19 summers at Friday Harbor, collecting hundreds of thousands of jellyfish to obtain enough of the elusive material for a full structural analysis. Until a way of making genetically engineered aequorin became available in the 1990s, Shimomura freely shared his carefully harvested stocks with laboratories the world over...
Remarkable.
He reminds me of another Japanese scientist who labored in obscurity on a difficult project, investing heavily his own time,
Shuji Nakamura (now at UC Santa Barbara).
One of my son's professors got his Ph.D. and did a post doc with Nakamura.