NNadir
NNadir's JournalI saw a great lecture today on how technology encodes racism.
Every winter the Princeton Plasma Physics Lab offers a series of lectures by scientists - most of them local, from the Plasma Physics Lab itself, Princeton University or Rutgers University, occasionally folks from other major universities - on various topics.
I originally went to expose my kids to science; they are men now, and still come when they can. I often see one of my good friends there; I used to take his son and he started coming on my own. If no one can join me, neither a son or my wife or my friend, I go alone. These lectures mean a lot to me.
Most of these talks end up as videos on the internet. Here is the archive, where you can watch many of them on line: Science On Saturday Archive.
The lecture I saw today was, Science on Saturday: "Will Robots Save Us… or Slay Us? Reimagining the Default Settings of Technology & Society."
You may think, looking at the title, as I did, that the topic would be sort of a reality based examination of the science fiction scenarios in which robots go wild and take over, displacing, and perhaps attacking and even eliminating humanity.
It turned out to be something quite different; something quite unexpected. It wasn't about the future, but about now.
Today's lecture was by Professor Ruha Benjamin, who is not a physicist, or chemist, or biologist but who is in Princeton's Department of African American Studies.
Dr. Benjamin is a very engaging speaker, very passionate, and is clearly a very profound thinker, and quite funny in a very trenchant and discerning way.
The topic was about how existing software, the software we use every day in our daily lives, how existing technological devices, everything from cell phone cameras to automatic soap dispensers in bathrooms, is coded to select for, well, white men.
The short video movie in the talk will make you laugh out loud - our audience did - while also breaking your heart; it broke mine when I realized what was happening. It should break your heart.
I wanted to be sure to note this lecture before becoming distracted by other things. It should be up on the SOS archive linked above by next week; last week's fascinating lecture already is.
I recommend this lecture highly. You cannot understand our world quite so well before watching it as you can after watching it.
Unlocking P(V): Reagents for chiral phosphorothioate synthesis of nucleic acid based therapeutics.
The paper I'll discuss in this post is this one:
Unlocking P(V): Reagents for chiral phosphorothioate synthesis
In many ways, I was a pretty stupid kid. I recall telling people that no one would ever make a protein drug.
Then they started being approved; and in recent years, they have come to dominate the pharmaceutical formulary, proving to be highly effective drugs.
In the past few years, we have begun to see drugs that are nucleic acids, designed to treat genetic diseases. They work quite well but tend to be very expensive because of the difficulty of synthesizing them on scale.
There was a time in my life that I was offered a job working on this sort of thing; I turned it down because I thought "that will never happen."
One of the pleasures of growing old is recognizing all the times you've been stupid.
One of the problems with DNA therapeutics is their kinetics; they tend to be broken down quickly by enzymatic processes in vivo. One mechanism for slowing the pharmacokinetics is to change the normal phosphate linkage group that builds the "sugar stairway" of nucleic acids to a thiophosphate, changing the oxygen in the phosphate to a sulfur.
An interesting thing about this is that doing so makes the phosphorous chiral, that is it is not superimposable on its mirror image. (If you have two hands, they are chiral, and the overwhelming majority of molecules other than water in your body are chiral.)
This allows for subtle control of the properties.
I just saw a lecture on this topic. I hadn't thought about "chiral phosphorous" in years, so it was cool to be reminded of this remarkable feature.
Mixed chirality can destroy a drugs effectiveness. An oligonucleotide that is 18 residues in length, for example, with 18 racemic (mixed chirality) thiophosphate groups would actually be 2^18 different compounds = 262,144 different compounds, only one of which would be effective, and many of which might be expected to be toxic.
From the paper:
Figure 1:

The caption:
Typically the stereochemistry of thiophosphate groups has been controlled using intermediates in which phosphorous in a reduced state, oxidation state (III) as opposed to the (V) state in DNA.
The authors challenged themselves to better control the stereochemistry of phosphorous to offer an inexpensive route to making these compounds:
The authors designed a new reagent utilizing limonene, a chiral compound found in great abundance in orange peels, limonene. (I have heard that the orange peels from the manufacture of orange juice can produce hundreds of metric tons of limonene.)
The reagent, which the authors call the "? reagent" is prepared as in this graphic:

(A) Stereochemical assignments. (B) Loading of nucleoside monomers. (C) Coupling to produce stereopure PS dinucleotides. R, TBDPS (tert-butyldiphenylsilyl). Loading: nucleoside [1 equivalent (equiv.)], ? reagent (1.3 equiv.), DBU (1.3 equiv.), MeCN, 25°C, 30 min. Coupling: nucleoside–P(V) (1.0 equiv.), coupling partner (2.0 equiv.), DBU (3.0 equiv.), MeCN, 25°C, 30 min.
Some more talk on the topic:
Figure 3:

The caption:
(A) Prior approach. (B) Stepwise and concerted macrocyclization using ?. *See (40) for the literature method. (C) Origin of observed stereochemistry in concerted protocol. PG, protecting group.
Automated nucleic acid synthesis using the reagent:
The caption:
(A) Crude HPLC trace of pentamer 23 (16 diastereoisomers) synthesized under standard P(III) automated conditions. (B) Crude HPLC trace of pentamer 23 (1 diastereoisomer) synthesized under unoptimized ? automated conditions. DMTr, dimethoxytrityl.
This sort of thing is an excellent tool for making drugs that are entirely unaffordable for the majority of the people who need them much, much cheaper, improving the quality of - and in some cases, saving - lives.
Have a nice evening.
A Broadening Definition in Fast Nuclear Reactor Design: A New Meaning for the Abbreviation "LMFR."
The paper I'll discuss in this brief post is this one: A Review of Molten Salt Reactor Kinetics Models (Wooten and Powers, NUCLEAR SCIENCE AND ENGINEERING · VOLUME 191 · 203–230 · SEPTEMBER 2018.)
The concept of "Molten Salt Nuclear Reactors" generally referred to as MSR's, has enjoyed a resurgence in recent years, and there are a plethora of start up companies devoted to them, some more likely to succeed than others. Even people who have a vague understanding that nuclear energy is the best (and in my opinion "only" ) tool for fighting climate change, but otherwise no deeper understanding of the technology, can generate enthusiasm for the "Liquid Thorium Molten Salt Reactor," about which they've heard, peripherally on the internet is "safer" and "generates less waste" than current nuclear reactors.
This is in spite of the fact that in comparison to all other forms of energy, the existing nuclear fleet is already "safer" and generates "less waste" than all other forms of energy. Combustion waste, approximately equally divided between "renewable" biofuels and dangerous fossil fuels - there is less difference between these two than people think - kills between six and seven million people per year. That is "unsafe." The quantity of combustion waste is in the tens of billions of tons per year, whereas used nuclear fuel accumulates at about ten thousand tons per year. Used nuclear fuel is generally a solid, easy to contain indefinitely, combustion waste is gaseous, impossible (on scale) to contain even briefly. All of the deaths associated with commercial nuclear technology utilized in a period of well more than 60 years, do not match the number of deaths that will take place in the next six days from combustion waste.
Molten salt reactors cannot be "more safe" than pressurized water reactors, because pressurized water reactors have half a century of experience of remarkably safe operation when measured against any other form of energy in terms of DALY/GWh. (DALY = Disability Adjusted Lost Years of life.) Any money spent to make nuclear reactors "safer" is wasted so long as fossil fuels and raw (and even processed) biomass exist, since they are both spectacularly "unsafe."
This said, Molten Salt Reactors are not really bad technologies; they have many features to recommend them, and other features that are may be less desirable, depending on design and chemistry of the working fluids. (The most commonly discussed working fluid is FLIBE, a eutectic mixture of lithium and beryllium fluorides.)
I've lost some of my initial strong enthusiasm for the concept of MSR's for various reasons, although I can't imagine any type of reactor, even an RBMK reactor of the Chernobyl type - no RBMK will ever be built again - that is dangerous as dangerous fossil fuels.
If people start building MSR's all over the world, so much the better for the world.
Most MSR's are built around the utilization of thorium based fuels, with the common isotope of thorium - 233Th - being a fertile rather than a fissile fuel. In recent years, I've been much more interested in plutonium - in the presence of fertile uranium 238 and/or 233 Th - because of certain remarkable features of plutonium metal across which I've come.
The MSR concept is largely based on a reactor built and operated at Oak Ridge National laboratory under the guidance of the great nuclear engineer Alvin Weinberg in the early 1960's. Another type of reactor - which has fascinated me in recent years - also operated around that time at Los Alamos, the LAMPRE, reactor, the "Los Alamos Molten Plutonium Reactor Experiment."
In the modern literature on nuclear reactors almost never discusses the LAMPRE concept, which involved the use of liquid plutonium metal as a eutectic with iron. Most of what I've learned about this reactor is from literature published in the 1960's, although there are some later papers that discuss the properties of liquid plutonium, for example, Properties of plutonium and its alloys for use as fast reactor fuels (Hecker and Stan, Journal of Nuclear Materials 383 (2008) 112–118).
The LAMPRE was not a reactor designed to have flowing fuel - as operated - but the paper above, is the first paper published in decades, to my knowledge - that refers to a new use of the abbreviation of LMFR, "liquid metal breeder reactor" - which refers in general to stationary solid nuclear fuels cooled by a liquid metal, generally and regrettably liquid sodium metal, in a new way, as a "liquid metal fueled reactor," a subtle and important difference.
To wit:
The history of MSRs begins with the Aircraft Reactor Experiment at Oak Ridge National Laboratory (ORNL) in the late 1950s and grows with the Molten Salt Reactor Experiment (MSRE), which was successfully operated with a full power of 8 MW(thermal) for more than 13 000 equivalent full-power hours with 233U, 235U, and 239Pu fuels at different points in time. No other MSR has been built since the MSRE project was closed in the late 1960s. However, MSR studies continued at ORNL, including the Molten Salt Breeder Reactor (MSBR) and the Denatured Molten Salt Reactor, and elsewhere in the world. In general, there was very little interest in developing MSRs from the 1970s through the 1990s. This changed in the late 1990s as interest in CFRs began growing in Europe. This momentum grew with the MOST project and continued through ALISIA, EVOL, and now SAMOFAR, resulting in the preconceptual Molten Salt Fast Reactor (MSFR). The Molten Salt Actinide Recycler and Transmuter (MOSART) design from Russia, the Chinese-designed Thorium Molten Salt Reactor–Liquid Fueled (TMSR-LF), the FUJI series of reactors out of Japan, and a host of proposed privately designed reactors have all additionally driven demand for modeling and simulation of CFRs.
The italics and bold are mine.
I like this new (to my knowledge) generalized abbreviation "CFR" which covers a broad range of possible reactor types.
For the record, my own thinking about liquid metal fueled does not involve flowing liquid fuels; I am interested in the potential of liquid fuels - owing to the metallurgical properties of liquid plutonium based fuels - to spontaneously reprocess some valuable and essential fission products. Some of these fission products were shown to be volatile at Chernobyl and Fukushima, where their uncontrolled volatilization accounted for their health risks, which despite being very real, are dwarfed by the health risks of volatile dangerous fossil fuel and biomass waste, risks humanity stupidly accepts continuously - under the normal operation of combustion equipment (as opposed to being in accidental settings) - without a whimper generally, and more generally with lip service not matched by action.
The paper focuses on MSR type reactors, but the point that it can be generalized is interesting.
The paper is basically about mathematical modeling.
For some flavor of its starting point, here's some more text:

where u is the fluid flow velocity and the number of groups is two with no upscattering.
"DNP" here refers to "delayed neutron precursor." Delayed neutrons allow for the control of nuclear reactors, if they flow out of a reactor before emitting neutrons, they can complicate reactivity.
In terms of generalization, for stationary liquid fuels this graphic from the paper interests me, as it touches on a problem about which I think quite often:

Some years ago, there was talk of a "nuclear renaissance" which looked backward to a time where humanity built more than 400 nuclear reactors in a period of about two decades, saving millions of lives that otherwise would have been lost to air pollution (dangerous combustion wastes). It didn't work out, owing to the willful destruction in the 1990's of intellectual and physical nuclear infrastructure by people who have a poor comprehension of basic facts, engineering, scientific or otherwise. This doesn't mean that we shouldn't look back to the life saving success of nuclear reactor engineering in the 1960's and 1970's, briefly continuing into the 1980's. These pressurized nuclear reactors were a spectacular success and they saved lives. The result of this failure of a nuclear renaissance has resulted in the rise of carbon dioxide concentrations by an unbelievable 45 ppm since January of 2000. The rate of the rate of increase, (the second derivative) is increasing, not decreasing as a result of this triumph of ignorance.
The triumph of ignorance is also rising, not falling. Part of this is political, but other factors are clearly involved.
Nevertheless, there are these pockets of knowledge - among them obscure nuclear engineering groups in national labs and universities - that function as monasteries did in the Middle Ages - that may bring future generations a better world, in spite of what we've done to them.
Language is a key to knowledge, and this subtle change of language where the letter "f" represents "flow" as opposed to - or in addition to - "fast" is a wonderful, if subtle, development.
I trust you're having a pleasant weekend.
The Decade's 1st Reading at the Mauna Loa CO2 Observatory, +3.45 ppm over 2019; 25.16 ppm over 2010.
The Mauna Loa carbon dioxide observatory which measures the concentration of the dangerous fossil fuel waste in the atmosphere daily, keeps a record of yearly increases and posts them on its website, both graphically and in a text menu on the right of the page where the data is recorded. The "official" yearly increase for a year generally is reported in February of the following year.
Annual Mean Growth Rate for Mauna Loa, Hawaii.
The record for all years since the Observatory opened in 1958 is 3.00 ppm over the previous year, recorded in 2016 over 2015.
The observatory also posts on its website - it can be found on the "data" page - a record of all weekly averages going back to 1976.
Somewhat obsessively I keep a spreadsheet of the weekly data, which I use to do calculations to record the dying of our atmosphere, a triumph of fear, dogma and ignorance that did not have to be, but nonetheless is. I note, with sadness and regret, that we on the left are not free of such fear ignorance and dogma, although I wish we were. We cannot, with justice, attribute this outcome to Ronald Reagan, George Bush the first and second, and Donald Trump. We bear responsibility, no matter how much we pat ourselves on the back for our insane, and frankly, delusional worship of so called "renewable energy."
So called "renewable energy" did not work, is not working, and will not work to address climate change. That's a fact.
Facts matter.
Here, in fact, is the first weekly data point recorded in the 2020's from the observatory:
Up-to-date weekly average CO2 at Mauna Loa:
Weekly value from 1 year ago: 409.94 ppm
Weekly value from 10 years ago: 388.21 ppm
Last updated: January 12, 2020
The reading, if you have not joined Greenpeace and thus can do simple arithmetic calculations, is 3.43 ppm higher than that recorded one year ago, and a somewhat startling 25.16 ppm higher than the weekly data point recorded in 2010.
This morning, I used the spreadsheet to calculate the average reading for all weekly data for 2019. It is 411.57 ppm. The same average, calculated in 2019 for 2018 was 408.56.
The Mauna Loa observatory calculates the "official" yearly increase figure using an average of the last weeks of the previous year and the first weeks of the following year. However, were they to use the average of all weekly readings, recorded throughout each year, this would mean that 2019 would set a new record, being the second year to reach the 3.00 ppm milestone at 3.01 ppm.
This data, although it constantly and monotonically is increasing owing to the accumulation of dangerous fossil fuel waste, is very much subject to a certain amount of statistical "noise" owing to the state of the biome. For example, until 2016 which came in at 3.00 ppm), 1998 was a record year at 2.98 ppm over 1997, owing to the South East Asian fires going out of control in a dry (in SE Asia) El Nino year. Many that went out of controlwere set to clear rain forest to plant palm oil plantations to meet the German demand for "renewable" biodiesel. Overall however, the average yearly increases in the 20th century were 1.54 ppm/year. In the 21st century, they are, thus far, averaging 2.41 ppm/year through 2018. The "official" 2019 figure will surely raise this average.
I pretty much expected, for much of this year, where "only" 5 of the weekly year to year increases were in the top 50 to come in among the 2,292 data points available at the Mauna Loa website, somewhere around 2.7 ppm, still an appalling figure, but not near 3.0. (2016 had 20 such readings that regrettably so qualified.) I think the recent surge for the 2019 figure is probably related to the Australian fires, which is not to say that things are "better than we think." These fires are examples of the feedback loop that is related to the accumulation of dangerous fossil fuel waste. Wait until "protecting the economy" strikes the Brazilian rain forest.
At the current rate represented by the 10 year figure for this week's reading of roughly 2.5 ppm/year (25.16/10) - I expect these figures will get worse, not better - we will reach 450 ppm by 2035. I'm sure that Bill McKibben, an advocate of the "renewable energy will save us" scheme, over at "350.org" will be very concerned. Maybe he should buy a "plug in" Prius. Electric cars will do nothing to save the world, but saving the world is not what counts. It's the thought that counts.
We have many things about which we should regard the Trump administration with absolute contempt: Ignorance, racism, incompetence, corruption, nepotism, blank and open immorality, disrespect for women, disrespect for the United States Constitution, betraying his country to Russian interests, war mongering, etc.
However, one reads criticisms of him for abandoning and criticizing so called "renewable energy." Again, it wouldn't matter if he supported with the same enthusiasm as the governing board of the Sierra club, which in a century's time has moved away from John Muir's establishing goal for the club, protecting wild spaces from development for things like "renewable energy" - in his time it was the lost goal of protecting Hetch Hetchy valley from the still existing Hetch Hetchy dam - to a policy of turning them into industrial parks for wind farms, solar thermal plants and the like, all of which will be rotting hulks requiring massive fleets of diesel trucks or diesel ships to haul them away in about 20 years time. So called "renewable energy" is a chimera.
If you don't believe me, don't worry, be happy.
As I remarked in my last post on this depressing and dire Mauna Loa data:
I personally consider such talk to be abysmally ignorant, of course, and an obvious statement that the people expressing such points are completely disinterested in the environmental issue of climate change as compared to their fear and ignorance connected with nuclear energy, but that's not my forum. I'm a scientist, not a cheerleader for the steel, aluminum, lanthanide and copper mining industries, nor the gas industry, industries on which so called "renewable energy" depends for its tortured (and hopefully short) existence.
Nuclear energy was, is, and always will be the only tool available to humanity capable of addressing climate change.
My impression is that I've been hearing all about how rapidly bird and bat grinding wind turbines are being installed, mostly written by simpleton anti-nukes, since I began writing here in 2002, when the reading on April 21, 2002 was 375.42 ppm.
All this jawboning about the wonderful growth of so called "renewable energy" has had no effect on climate change, is having no effect on climate change, and won't have any effect on climate change, but it's not climate change that counts: It's all that wonderful marketing showing pictures giant sleek wind turbines on steel posts that counts.
The reality - and I regret reference to reality in these times of triumph of the unreal - of what is happening is this:
In this century, world energy demand grew by 179.15 exajoules to 599.34 exajoules.
In this century, world gas demand grew by 50.33 exajoules to 137.03 exajoules.
In this century, the use of petroleum grew by 34.79 exajoules to 188.45 exajoules.
In this century, the use of coal grew by 63.22 exajoules to 159.98 exajoules.
In this century, the solar, wind, geothermal, and tidal energy on which people so cheerfully have bet the entire planetary atmosphere, stealing the future from all future generations, grew by 9.76 exajoules to 12.27 exajoules.
12.27 exajoules is slightly over 2% of the world energy demand.
Source: 2019 Edition of the World Energy Outlook Table 1.1 Page 38] (I have converted MTOE in the original table to the SI unit exajoules in this text.)
Don't worry; be happy. It's not your problem. It's the problem of every living thing that comes after us, including but not limited to human beings. We obviously couldn't care less.
Have a nice Sunday afternoon.
Libertango
Prokofiev: Piano Sonata No. 8, 1. Andante dolce
Solution of the Transport Equation in the Random Medium of High-Plutonium-Content HTR Lattice Cells.
Edit: Restored equation graphics.
The paper I'll discuss briefly in this post is this one: A New Collision Probability Approach for Solution of the Transport Equation in the Random Medium of High-Plutonium-Content HTR Lattice Cells (Indrajeet Singh,* S. B. Degweker,† and Anurag Gupta NUCLEAR SCIENCE AND ENGINEERING · VOLUME 189 · 101–119 · FEBRUARY 2018).
The problem of climate change has now advanced to a level such that future generations will be left with little choice but to attempt - it's uncertain whether this can actually be done, although it is possibly feasible - to reverse the addition of carbon dioxide to the atmosphere. It is notable that the sequestration of carbon dioxide - in the form of dangerous fossil fuels - took hundreds of millions of years, and thus any reversal will require intense engineering challenges, on a scale few of us can actually appreciate.
Let me give you a hint: Electric cars, solar cells, wind turbines and batteries won't cut it. These aren't even band-aids on the problem, never mind tourniquets; they're more like bringing styptic pencils to address the amputation of a leg.
The only feasible approach to address the engineering requirements will involve very high sustainable (24/7/365) temperatures which are only accessible using nuclear fuels.
The reactor described in this paper is not in a class that I personally favor, but it is capable of producing high temperatures. My problem with the reactor described herein is that it is designed to consume plutonium without generating new plutonium from depleted uranium. My feeling is that we must generate new plutonium at a rate much faster than it is currently being generated (largely in thermal spectrum reactors), which is roughly 100 MT per year. In order to completely eliminate all oil, gas, coal mining for energy, and, for that matter, uranium mining, and the vast quantities of copper, steel, aluminum mining required for the so called "renewable energy" industry - even at the trivial and useless levels at which it now exists - it is necessary to produce and fission (at 600 exajoules/year) 19 tons of plutonium per day. Therefore we cannot afford to consume plutonium without regenerating it.
Nevertheless, this paper addresses some interesting points about plutonium fuels that are worth contemplating:
From the introductory text, which discusses the type of reactor that dominates the current nuclear fleet, light water reactors, and, in a few places, notably India and Canada, the wonderfully neutron efficient heavy water reactors:
The paper assumes that all high temperature reactors are based on a type of fuel known as TRISO (for TRIStructural ISOtropic) fuel. In these fuels, actinides are suspended in a spherical carbon and silicon carbide matrix. It is not true that TRISO fueled reactors are the only option for producing high temperatures, but these types of reactors are generally the best known. These types of reactors are generally known as "pebble bed" reactors, early versions of which were more or less commercial failures with some exceptions, and in general they rely on the use of helium gas, which is not sustainable, world supplies of helium will have been depleted within a few generations: The gas released from party balloons boils off into space.
The paper describes the TRISO well enough as the introduction continues:
The authors note that the mean free path for uranium based fuels, including 233U obtained from thorium can be treated as a homogenous matrix, but this is not true for plutonium fuels:
...In this paper, we describe a new method developed for treating the thermal neutron transport in a random heterogeneous medium (which in the WIMS-D formalism contains the two large resonances of Pu that are not part of the resonance treatment method of this library). Themethod attempts to obtain exact expressions for the collision probabilities from one macroscopic mesh to another taking into account the random heterogeneous distribution of TRISO particles. Other than the statistical assumptions used to describe the medium, which fairly accurately describe the situations at hand, the method is exact.We present two independent derivations of the basic formulas. Both methods give identical expressions for the collision probabilities, which gives us confidence in the formulas so derived...
The paper, as one might expect, is mathematically heavy, some introductory flavoring:
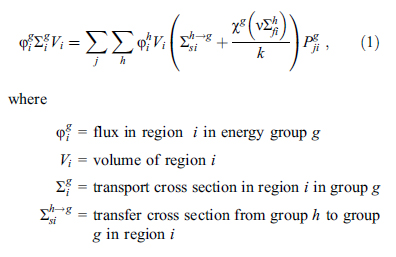

...ending up with this baby:

If this looks scary, don't worry be happy. The power of modern computers, which were not available at the dawn of the nuclear age, said dawn having produced some of the safest and cleanest energy devices ever commercialized (despite some very stupid press), specifically the pressurized water nuclear reactor, makes these sorts of calculations accessible.
This sort of thing is, of course, beyond the scope and understanding of dunderhead anti-nukes like, say, Amory Lovins, the Chief "Scientist" of the Rocky Mountain Institute, whose "Road Less Traveled" has become, regrettably, the "Road Most Traveled" with the result that as of this writing, we are seeing concentrations of the dangerous fossil fuel waste carbon dioxide in the atmosphere of over 413 ppm. This, as Mr. Lovins, says, is apparently "Winning the Oil End Game." Future generations will never forgive us for all this "winning," nor should they, since we took mindless assholes like Lovins seriously.
Anyway.
PMM is moderator to moderator probability. The reactor described herein is a moderated reactor which is designed to consume plutonium without regenerating it, something I find rather silly, since we need, again, more plutonium, not less of it. This reactor wastes valuable neutrons.
We do not need thermal (moderated) reactors to consume plutonium; we need fast (unmoderated) reactors to make more of it.
Speaking of plutonium, what I found interesting is the elemental composition of the fuel, specifically the isotopic distribution of the plutonium, shown in the following table:

This is "once through" plutonium, obtained from so called "nuclear waste" which is a silly term, since nothing that is extremely useful is actually "waste." At the very least what can be said for this type of putative reactor is that it at least utilizes plutonium as opposed to the very stupid idea of burying it as "waste."
Some other data relative to the Triso fuel composition and structure:

Some interesting data about the accumulation of americium and the interesting 242 isotope of curium, the latter representing a future source of helium, albeit, owing to the high energy density of nuclear fuels, nothing like the quantities we squander in this generation on balloons, at the expense of all future generations:

I hope you're enjoying the start of your weekend.
2019 Ended at the Mauna Loa CO2 observatory reporting a 3.54 ppm increase over the last 2018 reading
The Mauna Loa carbon dioxide observatory which measures the concentration of the dangerous fossil fuel waste in the atmosphere daily, keeps a record of yearly increases and posts them on its website, both graphically and in a text menu on the right of the page where the data is recorded. The "official" yearly increase for a year generally is reported in February of the following year.
Annual Mean Growth Rate for Mauna Loa, Hawaii.
Here, for convenience, is the graphic that can be accessed on that page:
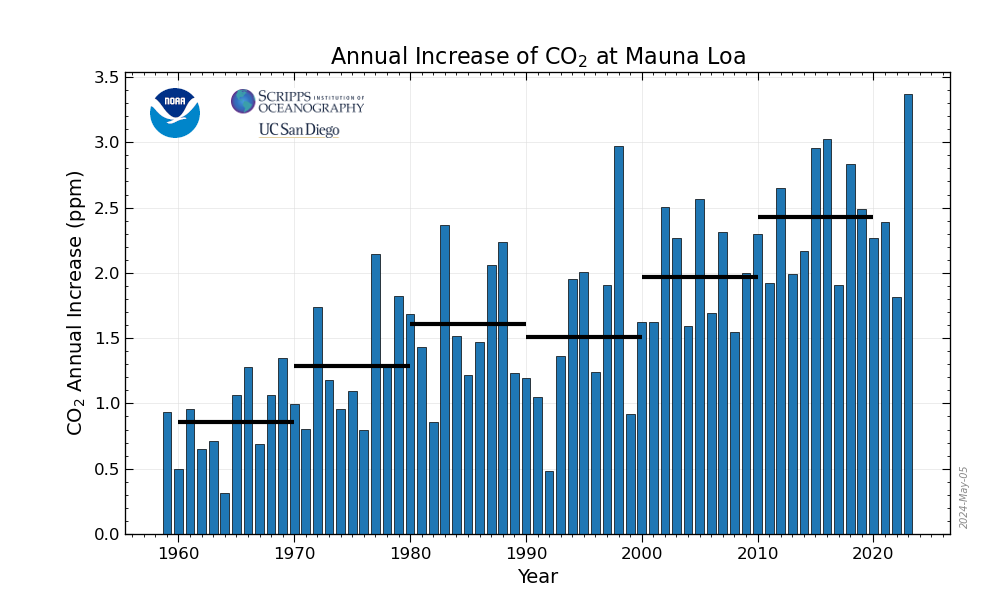
If you have good eye sight, you can make out that the first data point to reach the 3.00 ppm mark was in the decade just ended: It was recorded in 2016.
The designation of the end of the calendar year as marking the "official" increase is somewhat artificial; the concentration of carbon dioxide is sinusoidal each year, a sine wave's zero axis being more or less super imposed on a steadily rising axis that varies slightly from linear, as this graphic, also from the Mauna Loa website shows:

The actual maxima take place in late spring. In 2019, this maxima was reached during the week beginning May 12, 2019 when the concentration of the dangerous fossil fuel waste carbon dioxide was 415.39 ppm.
Still the end of the calendar year has some importance to "official readings" and it's useful to note what is happening at the end of the calendar year.
Here, again from the website is the "official data" for the week beginning December 29, 2019:
[link:Up-to-date weekly average CO2 at Mauna Loa|Up-to-date weekly average CO2 at Mauna Loa]
Weekly value from 1 year ago: 409.55 ppm
Weekly value from 10 years ago: 388.67 ppm
Last updated: January 5, 2020
Somewhat obsessively I keep a spreadsheet of the weekly data, which I use to do calculations to record the dying of our atmosphere, a triumph of fear, dogma and ignorance that did not have to be, but nonetheless is. I note, with sadness and regret, that we on the left are not free of such fear ignorance and dogma, although I wish we were. We cannot, with justice, attribute this outcome to Ronald Reagan, George Bush the first and second, and Donald Trump. We bear responsibility, no matter how much we pat ourselves on the back for our insane, and frankly, delusional worship of so called "renewable energy."
It is obvious that all the money thrown at so called "renewable energy" - the figures are in the trillions of dollars - did not work, are not working. I, for one, am absolutely and irrevocably certain that even more money, tens of trillions of dollars, will not work.
The reason is physics. The laws of physics are not determined by popular opinion, delusional or otherwise. They are independent of politics, and politicians ignore them at the peril of all humanity.
As of this writing, there have been 2,290 such weekly readings recorded at Mauna Loa, going back to 1975. If the increase recorded for the week of December 29, 2019 compared to the last reading of 2018, 3.54 ppm, had occurred before 2016, it would have easily been in the top 50 such readings. However 2016 recorded 20 of the top 50 readings as of this date. 2017 produced one such reading in the top 50, 2018, also one, five were recorded in 2019. In 2019, the ten year figures suggest that the current rate of carbon dioxide increases is between 2.4 - 2.5 ppm/year, the highest such rate recorded for any decade at Mauna Loa, going back to 1959. As it is, the 3.54 measurement for the week beginning December 29, 2019 is "only" the 70th highest out of 2,290 readings - tied with three other weeks - ever recorded.
The Mauna Loa observatory will not report 2019 as coming in at 3.54 ppm higher than 2018, thus shattering all previous records. Because of observational "noise" in the readings, the final "official" number is an average for the last weeks of the year averaged with the first weeks of the next year. The average for the last four weeks of these readings is 2.74 ppm higher than the same weeks of the previous year. Were this average to hold through the first several weeks of 2020, 2019 would "only" be the 4th worst observed in more than 60 years of collected data.
If the fact that this reading is 24.42 ppm higher than it was ten years ago bothers you, don't worry, be happy. You can read all about how wonderful things will be "by 2050" or "by 2100." Wind. Solar. Elon Musk. Tesla Car. And all that.
Don't worry. Be happy. Head on over to the E&E forum here and read all the joy expressed there for the German nuclear phase out.
I personally consider such talk to be abysmally ignorant, of course, and an obvious statement that the people expressing such points are completely disinterested in the environmental issue of climate change as compared to their fear and ignorance connected with nuclear energy, but that's not my forum. I'm a scientist, not a cheerleader for the steel, aluminum, lanthanide and copper mining industries, nor the gas industry, industries on which so called "renewable energy" depends for its tortured (and hopefully short) existence.
Nuclear energy was, is, and always will be the only tool available to humanity capable of addressing climate change.
My impression is that I've been hearing all about how rapidly bird and bat grinding wind turbines are being installed, mostly written by simpleton anti-nukes, since I began writing here in 2002, when the reading on April 21, 2002 was 375.42 ppm.
All this jawboning about the wonderful growth of so called "renewable energy" has had no effect on climate change, is having no effect on climate change, and won't have any effect on climate change, but it's not climate change that counts: It's all that wonderful marketing showing pictures giant sleek wind turbines on steel posts that counts.
The reality - and I regret reference to reality in these times of triumph of the unreal - of what is happening is this:
In this century, world energy demand grew by 179.15 exajoules to 599.34 exajoules.
In this century, world gas demand grew by 50.33 exajoules to 137.03 exajoules.
In this century, the use of petroleum grew by 34.79 exajoules to 188.45 exajoules.
In this century, the use of coal grew by 63.22 exajoules to 159.98 exajoules.
In this century, the solar, wind, geothermal, and tidal energy on which people so cheerfully have bet the entire planetary atmosphere, stealing the future from all future generations, grew by 9.76 exajoules to 12.27 exajoules.
12.27 exajoules is slightly over 2% of the world energy demand.
Source: 2019 Edition of the World Energy Outlook Table 1.1 Page 38] (I have converted MTOE in the original table to the SI unit exajoules in this text.)
Don't worry; be happy. It's not your problem. It's the problem of every living thing that comes after us, including but not limited to human beings. We obviously couldn't care less.
Have a pleasant work week.
Nanolaminated MAX Phases and Maxenes.
The paper I'll discuss in this post is this one: Element Replacement Approach by Reaction with Lewis Acidic Molten Salts to Synthesize Nanolaminated MAX Phases and MXenes (Huang et al, J. Am. Chem. Soc. 2019, 141, 11, 4730-4737)
This morning, in this space, I wrote a post about the phase diagram of supercritical sodium chloride, a surrogate for seawater, arguing that heating seawater to a supercritical state might help to address some important environmental issues, including but not limited to climate change. That post is here:
The Phase Diagram of Supercritical Water + Sodium Chloride
After writing it, I poked around in the literature a bit, and came across the paper to which this post points.
The MAX phases and Mxenes, materials technology brought to prominence by the Egyptian-American scientist Michel W. Barsoum - nobody asks me but I feel he should be a candidate for a Nobel Prize - are remarkable materials that kind of fit a "best of" category between ceramics and metals.
The paper cited above takes these materials to a new level that inspires some thinking about energy technologies that might utilize them.
Here is a graphic cartoon introducing the paper:

From the introduction:
So far, about 80 ternary MAX phases have been experimentally synthesized, with more continuously being studied, often guided by theory.(16,17) However, MAX phases with the A-site elements of late transition metal (e.g., Fe, Ni, Zn, and Pt), which are expected to exhibit diverse functional properties (e.g., magnetism and catalysis), are difficult to synthesize. For these late transition metals, their M-A intermetallics are usually more stable than the corresponding MAX phases at high synthesis temperatures, which means that the target MAX phases can hardly be achieved by a thermodynamic equilibrium process such as hot pressing (HP) and spark plasma sintering (SPS).
In 2017, Fashandi et al. synthesized MAX phases with noble-metal elements in the A site, obtained through a replacement reaction.(18,19) The replacement reaction was achieved by the replacement of Si by Au in the A layer of Ti3SiC2 at high annealing temperature with a thermodynamic driving force for the separation of Au and Si at moderate temperature, as determined from the Au–Si binary phase diagram. The formation of Ti3AuC2 is driven by an A-layer diffusion process. Its formation is preferred over competing phases (e.g., Au–Ti alloys), and the MAX phase can be obtained at a moderate temperature. Similarly, Yang et al. also synthesized Ti3SnC2 by a replacement reaction between the Al atom in Ti3AlC2 and SnO2,(20) although Ti3SnC2 can also be synthesized by a hot isostatic pressing (HIP) route.(21) Their work implies the feasibility of synthesizing novel MAX phases through replacement reactions.
It is worth noting that the synthesis of MAX phases by the A-site element exchange approach is similar to the preparation of MXenes by an A-site element etching process. Both are top-down routes that make modification of the A atom layer of pre-existing structure of the MAX phase, which involve the extraction of A-site atoms and the intercalation of new species (e.g., metallic atoms or functional terminals) at a particular lattice position. On the basis of this idea, we introduce here a general approach to synthesizing a series of novel nanolaminated MAX phases and MXenes based on the element exchange approach in the A-layer of the traditional MAX phase. The late transition-metal halides (e.g., ZnCl2 in this study) are so-called Lewis acids in their molten state.(22?25) These molten salts can produce strong electron-accepting ligands, which can thermodynamically react with the A element in the MAX phases. Simultaneously, certain types of atoms or ions can diffuse into the two-dimensional atomic plane and bond with the unsaturated Mn+1Xn sheet to form corresponding MAX phases or MXenes. The flexible selection of salt constituents can provide sufficient room to control the reaction temperature and the type of intercalated ions. In the present work, a variety of novel MAX phases (Ti3ZnC2, Ti2ZnC, Ti2ZnN, and V2ZnC) and MXenes (Ti3C2Cl2 and Ti2CCl2) were synthesized by elemental replacement in the A atomic plane of traditional MAX phases in ZnCl2 molten salts. The results indicate a general and controllable approach to synthesizing novel nanolaminated MAX phases and the derivation of halide-group-terminated MXenes from its respective parent MAX phase.
Some results from the paper:
Ti3ZnC2 was prepared by using the starting materials of Ti3AlC2 and ZnCl2 with a mole ratio of 1:1.5. Figure 1a shows the XRD patterns of initial phase Ti3AlC2 and final product Ti3ZnC2. Compared to Ti3AlC2, the XRD peaks of Ti3ZnC2 (e.g., (103), (104), (105), and (000l) peaks) are shifted toward lower angles, indicating a larger lattice constant caused by the replacement of Al atoms by Zn atoms. Note that the relative intensity of (0004), (0006) peaks increased while that of (0002) peaks decreased. This is caused by the change in structure factor by the replacement of the A atoms. According to a Rietveld refinement of the XRD pattern (Figure S2), the determined a and c lattice parameter of Ti3ZnC2 are 3.0937 and 18.7206 Å, which are larger than those of Ti3AlC2 (a = 3.080 Å, c = 18.415 Å).(3)
Figure 1:

The caption:
More text:
Figure 2:

The caption:
And now for something really, really, really, really cool, the chloroMAX phases:
A mixture of Ti3C2Cl2 MXene sheets and Zn spheres was obtained by using the starting materials of Ti3AlC2 and ZnCl2 with a mole ratio of 1:6. The SEM image (Figure 3a) and TEM images (Figure S5) of Ti3C2Cl2 show exfoliation along the basal planes. The corresponding EDS analysis (Figure S6) indicates that the elemental composition of MXene is Ti/C/Cl = 43.2:21.5:25.3 in atomic ratio, with small amounts of Zn (0.7 atom %), Al (2.9 atom %), and O(6.3 atom %). The presence of oxygen is reasonable because of prevailing O-containing compounds such as Al(OH)3, which is the hydrolysis product of AlCl3. Note that our theoretical calculation results indicate that the Cl terminations can strongly bond to the MXene surfaces but are not competitive with O-containing terminals.(40) Thus, a small part of the Cl terminations might be replaced by O-containing terminals during processes such as water washing, which could also contribute to the oxygen element detected on the surface. In addition, large Zn spheres can also be observed in the product, which can be easily distinguished from the MXenes (Figure S7)...
...A Ti 2p X-ray photoelectron spectrum of the as-reacted sample was shown in Figure 3d. The peak at 454.4 and 455.7 eV are assigned to the Ti–C (I) (2p3/2) and Ti–C (II) (2p3/2) bond.(41,42) The peak at 458.1 eV, attributing to a high valence Ti compound, is assigned to the Ti–Cl (2p3/2) bond.(43,44) Besides, the peaks at 460.3 eV, 461.8 and 464.1 eV are assigned to the Ti–C (I) (2p1/2), Ti–C (II) (2p1/2), and Ti–Cl (2p1/2) bonds, respectively...
Figure 3:

The caption:
The synthesis of these compounds is fairly straight forward, and involves heating the max phases in a ZnCl2 molten salt for zinc substitution, mixed KCl and NaCl molten salts for the chlorinated species.
The reaction progress was monitored by Energy Dispersive Spectroscopy (EDS) and X-ray diffraction (XRD).
Figure 4:

From the conclusion:
The formation of the Zn-MAX phases was achieved by a replacement reaction between Zn2+ and Al and subsequently the occupancy of Zn atoms in the A sites of the MAX phase. The formation of Zn-MAX phases indicates that such an exchange mechanism between traditional Al-MAX phases and the late transition metal halides might be a general approach for synthesizing some other unexplored MAX phases with functional A-site elements (such as magnetic element Fe). Late transition metal halides (e.g., ZnCl2), which have relatively low melting points and exhibit strong Lewis acidity in their molten state, seem to be ideal candidates for the replacement reaction. The acidic environment provided by the molten salts facilitates the extraction of the Al atoms from the A-atom plane in the MAX phase at a moderate temperature. The generation of the volatile Al halides in turn provides the driving force for the outward diffusion of the Al atom. Meanwhile, the liquid environment also facilitates the inward diffusion of replacement atoms, which finally promotes a thorough replacement reaction...
I have long wondered about the possibility of extending the MAX phase type structures across the periodic table beyond the early d-transition elements.
This is not my professional interest, but it is very, very, very, very cool, if esoteric.
I hope you're having a nice afternoon.
Mindless Anti-nuke Recognizes the Materials Problem with So Called "Renewable Energy."
The paper I'll discuss in this post is a "Policy Forum" article published in Science, this one: Sustainable minerals and metals for a low-carbon future (Benjamin Sovacool et al, Science Vol. 367, Issue 6473, pp. 30-33)
Despite all the cheering for so called "renewable energy," it is a fact that the trillions of dollars squandered on it has done nothing to address climate change.
This a reality, this is a fact, from the Mauna Loa CO2 observatory:
Up-to-date weekly average CO2 at Mauna Loa (Accessed 01/04/20)
Weekly value from 1 year ago: 409.24 ppm
Weekly value from 10 years ago: 388.17 ppm
Last updated: January 4, 2020
The rate of increases in the dangerous fossil fuel waste carbon dioxide, 2.4 ppm/year, over the last ten years is the highest such rate ever observed at the observatory over a ten year period, stretching back to the first such period ever recorded, that between 1959-1969 when the average rate was 0.85 ppm/year (based on yearly historical figures, not weekly year to year comparisons). As recently as the period between 1990-2000 the rate was 1.54 ppm/year, despite what was (then) a record year in 1998, when, owing to the out of control fires in Southeast Asia - fires set during an El Nino year to clear rain forests to plant palm oil plantations to make biodiesel for Germany's "renewable fuel portfolio standards" - the increase was 2.93 ppm in a single year.
Benjamin Sovacool is a prominent anti-nuke who has to be one of my least favorite commentators on energy and the environment, who nonetheless publishes frequently in scientific journals, about energy, even though he has, quite obviously, a very low appreciation of the physical sciences.
He has a BA in Philosophy; an MA and a Ph.D in something called "Science Policy" from Virginia Tech. These are not a physical science degrees; they are not an engineering degrees. They seem to be political science degrees.
Here is Doctor Sovacool's 2006 Phd Thesis: THE POWER PRODUCTION PARADOX: REVEALING THE SOCIO-TECHNICAL IMPEDIMENTS TO DISTRIBUTED GENERATION TECHNOLOGIES .
This is not a physical science thesis. The majority of the references in the thesis seem to be to "personal interviews." Sovacool sure can talk. Probably some of the people might have engineering degrees, but the transcripts of the interviews seem not to really involve much that is really hard engineering. It is possible that the "anonymous expert" who is a "high ranking executive" at a "large independent power producer" is an engineer, but basically the interviews I scanned from him consists of the same platitudes we've heard for half a century during which we have effectively done nothing. You do not need a Ph.D. to generate platitudes.
It has few references to physical science papers, although it does have at least one reference from blabber from Amory Lovins, who has consistently made loud predictions about energy and the environment since the 1970's, none of which have actually had much connection with what actually happened.
I have had one, and exactly one, personal interaction with Sovacool, when he logged onto a site where I was writing posts to inform me that his attitude toward nuclear energy is "complex."
This was in response to a criticism of his awful mentality, whereby his fantasies about "nuclear wastes" and "nuclear accidents" did very little to address the dangerous fossil fuel waste problem, which accounts for between 6 and 7 million deaths per year, as air pollution, and will certainly result in considerably more from climate change.
The worst nuclear disaster - an experimentally observed event at Chernobyl - will not result, in the next 100 years, as many deaths as will occur from air pollution in the next two days.
I responded to Sovacool's remarks on his "complexity" with derision.
The real problem with so called "renewable energy" is not that it has, at tremendous expense, failed to address climate change. The real problem is that the name for it is a lie. So called "renewable energy" is not "renewable" because its energy to mass ratio is absurdly small - which is why it was abandoned in the 19th century. While the technology may have changed since the 19th century, the mass intensity has not.
I have written a fair amount about that fact in this space. The subject of critical materials is garnering increasing attention, as well it should.
Some comments from Dr. Sovacool's Science paper:
Climate change mitigation will create new natural resource and supply chain opportunities and dilemmas, because substantial amounts of raw materials will be required to build new low-carbon energy devices and infrastructure (1). However, despite attempts at improved governance and better corporate management, procurement of many mineral and metal resources occurs in areas generally acknowledged for mismanagement, remains environmentally capricious, and, in some cases, is a source of conflict at the sites of resource extraction (2). These extractive and smelting industries have thus left a legacy in many parts of the world of environmental degradation, adverse impacts to public health, marginalized communities and workers, and biodiversity damage. We identify key sustainability challenges with practices used in industries that will supply the metals and minerals—including cobalt, copper, lithium, cadmium, and rare earth elements (REEs)—needed for technologies such as solar photovoltaics, batteries, electric vehicle (EV) motors, wind turbines, fuel cells, and nuclear reactors. We then propose four holistic recommendations to make mining and metal processing more sustainable and just and to make the mining and extractive industries more efficient and resilient.
Between 2015 and 2050, the global EV stock needs to jump from 1.2 million light-duty passenger cars to 965 million passenger cars, battery storage capacity needs to climb from 0.5 gigawatt-hour (GWh) to 12,380 GWh, and the amount of installed solar photovoltaic capacity must rise from 223 GW to more than 7100 GW (3). The materials and metals demanded by a low-carbon economy will be immense (4). One recent assessment concluded that expected demand for 14 metals—such as copper, cobalt, nickel, and lithium—central to the manufacturing of renewable energy, EV, fuel cell, and storage technologies will grow substantially in the next few decades (5). Another study projected increases in demand for materials between 2015 and 2060 of 87,000% for EV batteries, 1000% for wind power, and 3000% for solar cells and photovoltaics (6). Although they are only projections and subject to uncertainty, the World Bank put it concisely that “the clean energy transition will be significantly mineral intensive” (7) (see the figure).
Gee. I'm glad he found that out. I've been aware of it for well over a decade. In 2010, 2.1 trillion dollars ago in so called "renewable energy" "investments," for the week beginning January 3, 2010, the concentration of the dangerous fossil fuel waste carbon dioxide in the atmosphere was 388.06 ppm. Yesterday it was 413.22 ppm.
Recent Daily Average Mauna Loa CO2
January 02: Unavailable
January 01: 412.64 ppm
December 31: 413.20 ppm
December 30: 413.08 ppm
Last Updated: January 4, 2020
A graphic from Dr. Sovacool's paper, which I'm sure will be highly cited:

I'm not sure that any of this is accurate, by the way; it's not like we've developed superior magnets to the neodymium iron boride magnets or their dysprosium laced analogues for use in wind turbines. Perhaps Dr. Sovacool is counting on diesel powered ships to truck out to wind farms off the coast after the turbines have become waste in less than 20 years, and third world people to "recycle" these magnets, I don't know. (I have zero respect for his scientific knowledge.)
Other figures in real scientific literature are at odds with his graphics here.
He does pretend, in this paper, to care about the people who dig this stuff so we can all be "green."
Many of the minerals and metals needed for low-carbon technologies are considered “critical raw materials” or “technologically critical elements,” terms meant to capture the fact that they are not only of strategic or economic importance but also at higher risk of supply shortage or price volatility (8). But their mining can produce grave social risks. A majority of the world's cobalt, used in the most common battery chemistries for EVs and stationary electricity storage, is mined in the Democratic Republic of Congo (DRC) (see the map), a country struggling to recover from years of armed conflict. There, women and sometimes children often work in or around mines for less pay or status than their male and adult counterparts, without basic safety equipment (see the photo). Owing to a lack of preventative strategies and measures such as drilling with water and proper exhaust ventilation, many cobalt miners have extremely high levels of toxic metals in their body and are at risk of developing respiratory illness, heart disease, or cancer.
In addition, mining frequently results in severe environmental impacts and community dislocation. Moreover, metal production itself is energy intensive and difficult to decarbonize. Mining for copper, needed for electric wires and circuits and thin-film solar cells, and mining for lithium, used in batteries, has been criticized in Chile for depleting local groundwater resources across the Atacama Desert, destroying fragile ecosystems, and converting meadows and lagoons into salt flats. The extraction, crushing, refining, and processing of cadmium, a by-product of zinc mining, into compounds for rechargeable nickel cadmium batteries and thin-film photovoltaic modules that use cadmium telluride (CdTe) or cadmium sulfide semiconductors can pose risks such as groundwater or food contamination or worker exposure to hazardous chemicals, especially in the supply chains where elemental cadmium exposures are greatest. REEs, such as neodymium and the less common dysprosium, are needed for magnets in electric generators in wind turbines and motors in EVs, control rods for nuclear reactors, and the fluid catalysts for shale gas fracking. But REE extraction in China has resulted in chemical pollution from ammonium sulfate and ammonium chloride and tailings pollution that now threaten rural groundwater aquifers as well as rivers and streams. Several metals for green technologies are found as “companions” to other ores with differential value and unsustainable supply chains (9).
From my perspective, better late than never, even if in the 14 years since his Ph.D. thesis was published close to 100 million people have died from air pollution while he prattled on about "nuclear waste."
The United States has about 75,000 tons of used nuclear fuel, what in common parlance of people like Dr. Sovacool, is called "nuclear waste," although there is no intrinsic reason that it needs to be "waste." About 95% of this used nuclear fuel is unreacted uranium, slightly enriched with respect to natural uranium. About 1% of it is recoverable transuranium actinides, chiefly plutonium, and about 4% is recoverable (and valuable) fission products.
The world is consuming right now, with an ever increasing proportion of it coming from dangerous fossil fuels, about 600 exajoules of energy per year, up from less than 420 in 2000.
A kilogram of plutonium, completely fissioned, has about 80 trillion joules of recoverable energy. It follows that the unreacted uranium in used nuclear fuel, converted to plutonium in "breed and burn" reactors, represents, displacing all the dangerous oil, all the dangerous coal, all the dangerous natural gas, all the steel mines serving wind turbines, on the entire planet, at 600 exajoules per year, all of humanity's energy demand for about 9 and a half years. We have millions of tons of depleted isolated uranium available.
One million tons of uranium, converted to plutonium, is sufficient to supply all of humanity's current energy demand for well over a century. The ocean contains 4.5 billion tons of uranium, continuously being recycled out of Earth's mantle. The technology for extracting it is well known.
The energy content of this uranium is sufficient to last several orders of magnitude longer than human civilization has existed.
To this we may add the thorium partially isolated and dumped by lanthanide miners to serve the so called "renewable energy" industry.
I am unimpressed, that after nearly a decade and a half of spreading fear and ignorance, that Dr. Sovacool seems to be rethinking his rhetoric, although I very much doubt that this comment from his paper indicates that he has developed much of a shred of respect for humanity:
He still seems to believe that "distributed pollution" is better than concentrated pollution, and that the best way to serve the "40 million people" worldwide who dig this crap under appalling conditions, is to keep them poor to serve the "green" fantasies of bourgeois graduate students working on "social policy."
(He talks about making 965 million electric cars...what is he smoking over there in Brexit land?)
How about we educate and provide for those 40 million people and mine less?, not more in a pixilated scheme to chase after the "renewable energy" nirvana that has not come, is not here, and will not come? Crazy idea? Really?
You know what the problem really is? It's that incredibly poor people have not agreed to remain poor so that Americans can drive around in "green" Tesla cars. The per capita energy consumption of Chinese citizens is roughly a tenth that of an American, but we seem to have a problem with the fact that there are more Chinese than Americans, and that most of these Chinese are human beings who want to live decent lives. The same is true of Indians. And of Gabonese. And of Malians. And citizens of the "Democratic" "Republic" of the Congo.
The key to sustainable energy is an extraordinarily high energy to mass ratio, one higher than the high ratio available from dangerous fossil fuels, which we continue to use - in our short term mentality - because of their high mass to energy ratio. Only nuclear fuels, actinides and (if it ever becomes commercially feasible to use it) tritium and deuterium have such ratios exceeding those of dangerous fossil fuels which, must be banned if there is really any concern for ethics. The key to sustainability is also contiguous, in my opinion, with human development goals, decent lives for all the citizens of the world.
It's a matter of ethics but so far as energy it is also a matter of physics and applied physics (engineering), the laws of which are not subject to repeal by legislatures, even those legislatures goaded by "social scientists."
If I sound angry, it's probably because I am.
I hope you're having a happy New Year thus far.
Profile Information
Gender: MaleCurrent location: New Jersey
Member since: 2002
Number of posts: 33,518