NNadir
NNadir's JournalIs the atmosphere momentarily too unstable to measure?
In analytical chemistry we have certain rules by which we test the validity of data, one being precision, which is the spread of data - the width of the spread - i.e. reproducibility over multiple measurements.
The Mauna Loa CO2 Observatory uses precision determinations to qualify its data; it's very high end analytical chemistry using a laser light absorbance technique known as "ring down spectroscopy." Over the last few days as of this writing, the daily measurements have been rated "unavailable" because of wide spreads in the data, with some hourly readings measuring over 430 ppm.
It's best to show this graphically from the website:
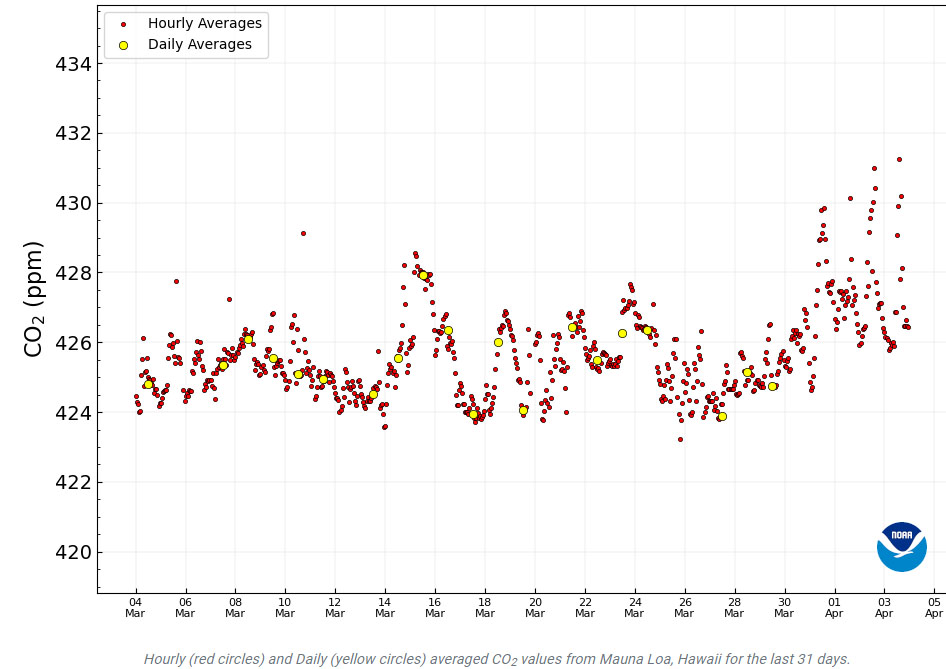
Recent Daily Average Mauna Loa CO2 (Accessed O4/04/2024)
A lack of precision can be instrumental, and often it is. There is a whole science of measurement designed to identify errors. However if one is analyzing an unstable system, precision can suffer because the variance in the data is real.
We are in new territory at the Mauna Loa CO2 Observatory; what I'm seeing there is blowing my mind. Over many years of accessing the data, I've never seen anything quite like 2024, as I noted here:
2024's Unprecedented Terror At the Mauna Loa CO2 Observatory Continues.
One hopes that the issue in these daily readings is instrumental and that it is not a function of real instability.
We'll see.
Since I joined DU in November 2002 I've been hearing, whenever climate issues are discussed, how a reactionary program of returning to the early 19th century in placing our energy sources dependent on the weather, so called "renewable energy" - precisely at the time we destabilized the weather with wishful thinking, selective attention, and soothsaying - the concentration of the dangerous fossil fuel waste carbon dioxide in the planetary atmosphere has risen by 52.08 ppm. In this century as a whole, beginning before I joined DU, the carbon dioxide concentrations have risen by 56.68 ppm. (These numbers are based on weekly averages at the Mauna Loa CO2 Observatory.)
While there is noise in the measurements on an hourly, daily, weekly, monthly and annual data, the analytical "rules" are designed to prevent inaccuracies inherent in owing to the methods themselves but clearly, the overall trends are surely real. We are not doing anything meaningful to address climate change other than jawboning insipidly.
That, at least, is an accurate statement.
I'm reading through Eleanor Roosevelt's Correspondence with President Harry Truman.
Here's the book:
Eleanor and Harry: The Correspondence of Eleanor Roosevelt and Harry S. Truman (Lisa Drew Books)
The forward is (appropriately) from Gloria Steinem.
I have always regarded Ms. Roosevelt as the premier Democrat of the 20th century (maybe of any century), rating above her husband in terms of the values that make me a Democrat.
Reading the detailed and wise advice she gave the President in long correspondence, and his appreciation of that advice in response, the clarity of her mind, the decency, to my mind, make her far and away the greatest "first lady," in our history, a most remarkable American, and indeed on the level of our finest Presidents. I have no doubt that Mr. Truman's outstanding Presidency profited from her very real influence.
Among first ladies, only Abagail Adams, Michelle Obama, and Hillary Clinton can compare on the point of raw intellect and a deep sense of decency.
What a remarkable American she was!! To paraphrase Willie the Shake, "all in all, we will not see her like again."
Teaching to support religious ignorance in public schools legalized in West Virginia.
A news item from the current issue of Science.
West Virginia opens the door to teaching intelligent design
Subtitle:
Science 18 MAR 20243:30 PM ETBYJEFFREY MERVIS
The bill, which the state’s governor is expected to sign before the end of the month, is the latest example of what Nicholas Matzke, an evolution educator at the University of Auckland, has called “legislation that avoids mentioning creationism in any of its varieties but advances creationist antievolutionism.” It comes nearly 2 decades after Jones, now president of Dickinson College, told the Dover, Pennsylvania, school board that its policy on teaching ID violated the so-called “establishment” clause of the U.S. Constitution that bans the government from taking action favoring any religion.
Last year, the West Virginia Senate approved a bill that would have specifically allowed teachers to talk about ID “as a theory of how the universe and humanity came to exist.” But the measure died in the House of Delegates. This year, State Senator Amy Grady (R) reintroduced the bill but revised it to remove the words “intelligent design.” Her colleagues approved it in late January and the House followed suit on 9 March, sending the bill to Republican Governor Jim Justice...
Is the US Department of Energy Anti-German?
It would seem so, at least where energy policy is concerned, since Germany replaced nuclear energy with coal, killing people and the planet.
From the US Department of Energy:
Coal-to-nuclear transitions could dramatically increase the supply of reliable, clean electricity to the grid and make progress toward the nation’s goal of net-zero emissions by 2050.
“As we work to transition to a net-zero economy, it’s absolutely essential that we provide resources to energy communities and coal workers who have helped our nation’s energy system for decades,” said Assistant Secretary for Nuclear Energy Dr. Kathryn Huff. “This is a core promise of the Biden-Harris Administration: to deliver place-based solutions and ensure an equitable energy transition that does not leave energy communities behind.”
This information guide builds on DOE’s 2022 study that found hundreds of U.S. coal power plant sites across the country could be converted to nuclear power plant sites. DOE’s information guide offers communities a high-level look at the economic impacts, workforce transition considerations...
DOE Study Finds Replacing Coal Plants with Nuclear Plants Could Bring Hundreds More Local Jobs and Millions in Added Income and Revenue to Energy Communities
More important, to my mind, than providing jobs, as far as advancing nuclear energy is concerned - something in which the Biden administration is producing the best record of any administration in half a century - is saving human lives, but yes, building nuclear infrastructure back better will create very high tech jobs.
For full disclosure, a member of my family is sure to hold one of those jobs.
EST: Chinese Hydrogen Production Is Making Climate Change Worse.
Discussing climate change around here is often an exercise in delusion; whatever it is we think we're doing is making things worse, not better, and it's making things worse faster than ever.
2024's Unprecedented Terror At the Mauna Loa CO2 Observatory Continues. In all the years I've monitored the Mauna Loa CO2 Observatory, I've never seen anything like 2024.
We have around here, a fossil fuel sales team, including an apparent bot, working to rebrand fossil fuels as "hydrogen," this to encourage fossil fuel sales by greenwashing them, often juxtaposing, in a bait and switch fashion, advertising graphics of useless solar farms next to slick similar pictures of hydrogen stations in China to promote the lie that hydrogen is made using so called "renewable energy," which despite trillions of dollars and worldwide enthusiasm has done nothing to address the use of dangerous fossil fuels, and has, in fact, entrenched them.
I have noted that in China, as elsewhere, in my own words, just as is the case everywhere else on this planet, hydrogen is made overwhelmingly by the use of fossil fuels, including the tiny amounts made by using grid electricity, with exergy destruction, the term "exergy destruction" being a thermodynamic term for "wasted energy," i.e. low energy efficiency, waste, a consequence of the inviolable 2nd law of thermodynamics.
A Giant Climate Lie: When they're selling hydrogen, what they're really selling is fossil fuels.
Here's a publication by Chinese scientists, more or less saying exactly what I've been saying:
Subsidizing Grid-Based Electrolytic Hydrogen Will Increase Greenhouse Gas Emissions in Coal Dominated Power Systems Liqun Peng, Yang Guo, Shangwei Liu, Gang He, and Denise L. Mauzerall Environmental Science & Technology 2024 58 (12), 5187-5195
The text is clear enough.
From the introductory text:
Electrolytic hydrogen can be categorized by its electricity source: grid-based hydrogen generated using electricity from the power grid and renewable-based hydrogen generated directly from renewable electricity. Grid-based hydrogen is cheaper than renewable-based hydrogen in most provinces, requiring lower subsidies for its development. However, grid-based electricity generation relies heavily on coal and, thus, has substantial GHG emissions. Subsidizing grid-based hydrogen production would likely increase GHG emissions relative to coal-based hydrogen production, whereas hydrogen directly generated from renewable energy has minimal GHG emissions.
China aims to reach peak carbon emissions by 2030 and to achieve net-zero carbon emissions by 2060. To minimize cumulative carbon emissions, accelerating the transition to decarbonized hydrogen production is crucial. However, high production costs are a significant barrier to the adoption of clean electrolytic hydrogen. Currently, renewable-based hydrogen and grid-based hydrogen cost 2–6 times and 1.6–3 times more than coal- or SMR-based hydrogen, respectively. (2,5?7) Until renewable-based hydrogen becomes cost-competitive, large-scale development of hydrogen is likely to increase GHG emissions by expanding fossil- and grid-based electrolytic hydrogen production. To rapidly decarbonize the hydrogen production process, it is essential to accelerate the shift from fossil- to renewable-based hydrogen. While some provincial governments (e.g., Inner Mongolia and Gansu) have established hydrogen production goals, their plans lack a specific focus on renewable-based electrolytic hydrogen. Moreover, there is little research at the provincial level that compares life cycle GHG emissions and levelized costs of hydrogen (LCOH2) across all hydrogen production technologies. Our work provides valuable insights into the trade-offs between subsidies and GHG emissions in the development of the hydrogen industry at the provincial level in China.
Subsidies play a significant role in developing emerging technologies. To accelerate the electrolytic hydrogen transition, subsidies on hydrogen-related devices and hydrogen used for transportation have been deployed in different regions as pilot projects. (8,9) Since renewable-based hydrogen costs 2–6 times more than coal- or SMR-based hydrogen, (2,4,5) greater subsidies are required to make renewable hydrogen cost competitive and to drive commercial production...
I added the bold, underline and italics. I note that we have lots of people around here who embrace consumer balderdash about cost all the time, particularly with respect to the alleged cost of nuclear energy, because investments in nuclear energy will accrue benefits to future generations and not the assholes whining about pennies in their pockets now. Notably, the fossil fuel sales people and sales bots around here embrace antinuke rhetoric enthusiastically, which is unsurprising, because, well, the reason they greenwash fossil fuels is to sell them, not because its hard to sell fossil fuels, but because they want to extend sales indefinitely.
The argument these people make is that nuclear energy is "too expensive" but climate change isn't "too expensive."
In the State of New York, led by the actor Mark Ruffalo, whose environmental science qualifications seem to include being filmed having simulated (or perhaps real) sex with the actress Emma Stone, this decision to indicate that climate change isn't "too expensive" or "too dangerous" but nuclear power is "too dangerous" and "too expensive" has been described in the newspaper The Guardian, hardly a source of "right wing talking points."
A nuclear plant’s closure was hailed as a green win. Then emissions went up.
One would need an education in a subject other than acting in soft porn films to know what "green" means, that it might have something to do with climate change which is a serious matter, something deaths from radiation at Indian Point never was.
In defense of China, and its efforts to address climate change, I note that China has the best operating nuclear power plant construction infrastructure in place right now, and while it is squandering money, vast sums, on so called "renewable energy" it is also building nuclear plants at a pace not seen anywhere on Earth since the 1960s and 1970s in the United States and France in the 1980's. China will surpass France as one of the largest producers of nuclear power in short order, and in slightly longer order, the United States, still the world's leader in nuclear power production, owing to a scientific, engineering, and industrial infrastructure that built nuclear power plants nearly half a century ago, leaving a gift to our generation.
I note that making hydrogen from nuclear electricity is just as wasteful as making it from any form of electricity, as the cited EST article points out. One hears of high "faradaic efficiencies" but the more important "thermodynamic efficiency" is often buried in texts and requires calculation from the over voltages. (It isn't pretty.)
Like all "hydrogen will save us" marketing that is in effect the marketing of dangerous fossil fuels, there's all kinds of soothsaying in the paper cited at the outset about what "could" be done with hydrogen to address the appalling effort to accelerate the destruction of the planetary atmosphere that existing hydrogen technology involves everywhere on the planet. That appalling effort is succeeding spectacularly since the acceleration is underway, the first derivative, second derivative and third derivative with respect to time of carbon dioxide accumulations in the planetary atmosphere are all positive. (When I integrate the second derivative twice to obtain a crude quadratic, substitute the boundary conditions represented by the present data, and solve for 500 ppm concentrations of the dangerous fossil fuel waste carbon dioxide in the planetary atmosphere, it seems, by this crude model, we'll hit that figure around 2046.)
One of the appalling ideas advanced (in the soothsaying part) in this otherwise interesting paper describing current reality is to use existing gas pipelines as hydrogen pipe lines from some magical so called "renewable energy" nirvana off in some Chinese wilderness somewhere. I would suggest that the people writing this sort of thing take an introductory course in metallurgy to learn what the term "hydrogen embrittlement" means.
Anyway...
If you're a Christian who embraces the phenomenological bit about people rising from the dead, have a very happy Easter. I'm not sure a stable planet can be resurrected, but have a happy holiday anyway irrespective of what I think.
My $300 two year old watch stopped, and I went to get the battery changed where I bought it.
I was informed at the store that it would need to be sent out if I wanted it fixed for a battery replacement but the problem was easily addressed by leaving it in the sunlight for 10 days.
I had no idea of this, but it appears that it's solar powered; the watch face is a solar cell.
I wear long sleeved shirts at my job.
This explains why it would seem to restart and then fail.
This sort of thing is in store for the entire planet, by the way, but don't worry, be happy.
Talking About My Generation.
With the atmosphere collapsing under the rule of fear and ignorance coupled with unholy dollops of wishful thinking and faith based nonsense, I'm out of things to say here, but that aside, I couldn't resist noting this book commentary from Publisher's Weekly across which I stumbled this morning:
Triumph of the Yuppies: America, the Eighties, and the Creation of an Unequal Nation
Vaulting ambition, passionate consumerism, and a business culture that threw workers under the bus are among the achievements of the yuppie generation, according to this penetrating study. Journalist McGrath (MTV) charts the trajectory of the “young urban professional” cohort who protested in the 1960s, “found themselves” in the ’70s, and went to Wall Street in the ’80s. Their impact, he notes, was far-reaching: they gentrified America’s cities with chic restaurants and shops, driving up rents; fetishized luxury brands and artisanal foods like Cuisinart, BMW, and Perrier; forged a self-congratulatory concept of success by flaunting their advanced degrees and endless work hours; and embraced Ronald Reagan’s vision of unfettered corporate capitalism. McGrath hangs his analysis around portraits of colorful personalities like Jerry Rubin, a former lefty radical who started a company that hosted business-networking parties, and barbed accounts of the yuppies’ oft-satirized quirks, from their dreary jargon to their reflexive crassness (“I’ve got a fast-track career.... And now I need a faster-track relationship,” he quotes one saying as he dumped his wife for a coworker). He also reckons the cost of the yuppie-administered 1980s economy with haunting profiles of rust belt towns like Youngstown, Ohio, that lost millions of manufacturing jobs. It’s a beguiling look at an era that inaugurated an ever-widening rift between a self-satisfied elite and a resentful working class. (June)
Sounds about right to me...
Apologies to the two surviving old men in The Who who sang "I hope I die before I get old..." but didn't.
Forget 'Oppenheimer' -- nuclear power is having its moment in Washington
Forget ‘Oppenheimer’ — nuclear power is having its moment in WashingtonSubtitle:
Politico, By JAMES BIKALES and CATHERINE MOREHOUSE
03/11/2024 02:18 PM EDT
Some Excerpts:
Freshly passed legislation and new rules from the Biden administration are putting teeth behind a renewed bipartisan push for nuclear power, which has suffered major setbacks in recent decades despite advocates’ repeated predictions over the years that the industry was on the brink of a renaissance.
The bipartisan support for nuclear power in today’s divided Washington has been well-documented, especially given its potential importance as a carbon-free energy source that can aid the fight against climate change. But that hasn’t necessarily translated to concrete policy progress for an industry that has hit hurdle after hurdle...
... President Joe Biden on Friday signed into law the fiscal 2024 spending bill for the Energy Department and several other agencies, which includes $2.7 billion for DOE to boost domestic uranium production. The funding was a rare point of widespread agreement between Democrats and Republicans, and exceeds a request for $2.2 billion the White House made last year — using money repurposed from the bipartisan infrastructure law.
The funds are on top of new authorities for domestic uranium production that Congress provided in the Nuclear Fuel Security Act, which made it into last year’s National Defense Authorization Act. That law created new programs at DOE to provide incentives for production of the fuel — known as high-assay, low-enriched uranium — that will be critical for advanced reactors but is largely produced in Russia. DOE has moved quickly to implement the law, and on Friday moved to begin evaluating proposals for HALEU enrichment contracts...
A "tweet" from Energy Secretary Granholm:
We are in a transformational moment where we can build advanced nuclear reactors at a pace and scale not seen since the 1970s.
Let's leverage
@POTUS
' once-in-a-generation investments and make 2024 a year of action for nuclear.
It is all "too little, too late," with respect to climate change, but it is our last, best hope to save what is left to save and perhaps restore some of what can be restored.
Enjoy the work week.
2024's Unprecedented Terror At the Mauna Loa CO2 Observatory Continues.
As I've indicated repeatedly in my DU writings, somewhat obsessively I keep a spreadsheet of the data at the Mauna Loa Carbon Dioxide Observatory, which I use to do calculations to record the dying of our atmosphere, a triumph of fear, dogma and ignorance that did not have to be, but nonetheless is, a fact.
Facts matter.
When writing these depressing repeating posts about new records being set, reminiscent, over the years, to the ticking of a clock at a deathwatch, I often repeat some of the language from a previous post on this awful series, as I am doing here with some modifications. It saves time.
This is a regrettable add on to my post of last week, which referred to the regrettable add on to the post of the previous week which in turn referred to the post of the week before that:
At the Mauna Loa CO2 Observatory, the 2024 Terror Continues.
I've been at this for a long time, and I've never seen anything quite like the beginning of 2024, and the shock continues week after week of this year.
Let's cut to the chase with some statistics on what is already shaping up to be a mind blowing disaster in terms of accumulations of the dangerous fossil fuel waste carbon dioxide.
The data from week 10 of 2024, which, if it is continues as it has been going through the first 10 weeks, will certainly go down in history as a landmark disaster:
Weekly value from 1 year ago: 420.08 ppm
Weekly value from 10 years ago: 400.31 ppm
Last updated: March 17, 2024
Weekly average CO2 at Mauna Loa (Accessed 3/17/2024)
This week's increase from week 10 of 2023 compared to week 10 of 2024 is 5.66 ppm.
There are 2511 weekly data points recorded on the Mauna Loa Observatory's pages; this is the 2nd highest increase over the same week of the previous year ever recorded. It is one of only 26 readings out of 2511 to exceed an increase of 4.00 ppm over the reading of the same week of the previous year. Of the fifty highest such readings out of 2511, six have taken place in the first 10 weeks of 2024, included the three highest ever recorded included, 5.75 ppm higher in week 5 of 2024 in comparison to week 5 of 2023, and 5.53 higher in week 7 of 2024 compared to week 7 of 2023.
The year is still young.
Week 5 and week 7, and now week 10, of 2024 represent three of only four such week to week comparators with readings of the previous year to exceed a 5.00 ppm increase. The only other such an increase to exceed 5.00 ppm occurred in 2016, the previous "worst year ever" in CO2 accumulations, 5.04 ppm recorded in the week beginning July 31, 2016, week 28 when compared with week 28 of 2015.
Of the top 50 highest readings of the difference between weeks of the year with those of the previous year out of the 2511 such data points, 15 have taken place in the last 5 years, 36 in the last 10 years, and 43 in this century. Of the seven readings from the 20th century, four occurred in 1998, when huge stretches of the Malaysian and Indonesian rainforests caught fire when slash and burn fires designed to add palm oil plantations to satisfy the demand for "renewable" biodiesel for German cars and trucks as part of their "renewable energy portfolio" went out of control.
From 1988 to 1998, the record for the highest such comparator was a reading of a 3.91 ppm increase recorded in week 34 of 1988, the week beginning in August 21, 1988, as compared to week 34 of 1987. It is now the 30th highest such reading.
In the week to week comparisons with readings 10 years ago, week 10 of 2024 compared to week 10 of 2014 the reading is 25.47 ppm higher. The top four of all such ten year comparators all occurred in the first ten weeks of 2024. All of the top fifty such readings have taken place since 2019. Seven of these are from the young year of 2024.
A 52 week running average of 10 year week to week comparators has now reached 24.78 ppm/10 years, the highest level ever recorded. In the tenth week of 2001, that running average was 15.31 ppm/10 years.
Since the first week of the year 2000, the week beginning January 2 of that year, the increase in the concentration of the dangerous fossil fuel waste carbon dioxide in the collapsing planetary atmosphere has been 57.08 ppm.
Since I joined DU in late November 2022, when, then as now, the main issue on my mind is the relationship between energy and the environment, the increase in the concentration of the dangerous fossil fuel waste CO2 has been 53.21 ppm.
In reaction to these obscene numbers, I've heard endlessly from people for whom I have no intellectual, moral, scientific, professional or educational respect, all about Chernobyl, and then about Fukushima, as if these fucking trivial events matter in comparison to an entire planet in flames. The appalling indecency of these tiresome, if popular, fools, to my mind, is on a Trumpian scale:
The number of people killed by air pollution at a rate of 6 to 7 million people per year since Chernobyl in April of 1986 is between 230 million and 270 million human beings; since March of 2011, the death toll from the same cause, air pollution, has been between 80 and 90 million human beings since the disgusting distraction obsession with Fukushima began, continuing right up to this day, working to advance the use of dangerous fossil fuels, about which the mindless antinuke community of intellectual and moral Lilliputians could not care less. These death tolls do not include the vast death tolls associated with climate change, extreme weather, heat stroke, etc...
Global burden of 87 risk factors in 204 countries and territories, 1990–2019: a systematic analysis for the Global Burden of Disease Study 2019 (Lancet Volume 396, Issue 10258, 17–23 October 2020, Pages 1223-1249)
Climate change was already killing people around the time I started writing here:
Death toll exceeded 70,000 in Europe during the summer of 2003 Robine et al., Comptes Rendus Biologie, 331 (2008) 2, 171-178.
Things are unambiguously much worse in 2024 than they were in 2003.
My anger is difficult to contain particularly with respect to the, again, Trumpian cretins who want to tell me that this is OK because they have barely enough brain cells left to mutter "Fukushima" or "Chernobyl" as if these words mattered on any scale of human decency.
No decency, zero, zilch, diddly squat, nix, nothing, among this set, none!
Once in this space I mused about the mythical - or not so metaphorically mythical - character of Cassandra, who was blessed by the gods with prescience but was required by them to always proclaim the truth. Her reward for always correctly predicting the truth was to be raped and murdered.
So be it.
Kill the messenger.
Of course, as opposed to the time of the Trojan war, we now understand powerfully the predictive power of numbers. The numbers above should speak to anyone with a modicum, even a mote, of intelligence. If they don't, don't bother to tell me about it. I'm depressed enough at the end of my life to see the planet in flames because of fear, ignorance, egged on by advertising to whip up insipid and highly selective attention. I really don't need to contemplate another in a long series of oppressive idiots I've encountered over the years, here and elsewhere.
Spare me.
The other stuff I say while reporting on this 2024 disaster, the one going under the radar while we all whine and carry on about things other than a planet in flames, our planet:
As I've been reporting over the years in various contexts, the concentrations of the dangerous fossil fuel waste carbon dioxide which is killing the planet fluctuate sinusoidally over the year, with the rough sine wave superimposed on a roughly quadratic axis:
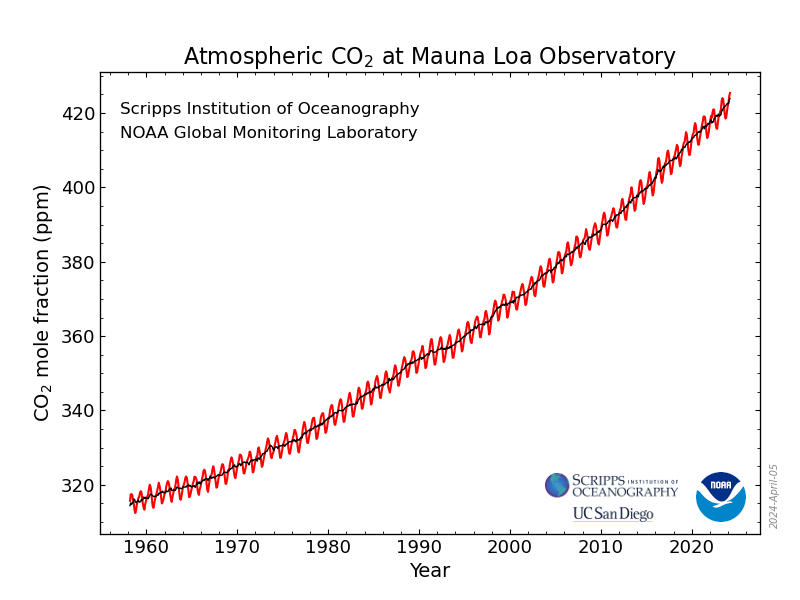
Monthly Average Mauna Loa CO2
There is some statistical noise in these readings, but the overall trends are clear enough, inescapable, dire, terrifying, even as they are largely ignored or swept from attention by cheap diversions:
In spite of these ever worsening and ever more astounding numbers - people lie to each other and to themselves but numbers don't lie - you will still find people mindlessly cheering for fantasies about bourgeois toys that do nothing to address climate change, be they electric cars, solar cells and/or wind turbines, all of which are exercises in promoting the use of fossil fuels, the destruction of wilderness, and the demand for mining. We also have people here and elsewhere selling fossil fuels by rebranding them as "hydrogen," the production of hydrogen, which overwhelmingly made from fossil fuels, involving exergy destruction and thus driving climate change faster along with all of the other public fantasies.
The big lie people tell themselves and each other that these pixilated reactionary schemes, electric cars, solar cells, wind turbines, hydrogen blah, blah, blah is "doing something" about climate change. This is nonsense. That it is nonsense is clearly shown, again, by the numbers. The reactionary scheme of carrying on about so called "renewable energy" that led us here was never about climate change or any other environmental issue and the claim that it is is an afterthought. It was always about attacking the only realistic alternative to fossil fuels, nuclear energy.
The antinukes won and humanity, and in general, the rest of the biosphere lost.
We're clueless.
A paper addressing the idea that electric cars are "green."
Let's be clear: People lie to themselves and they lie to each other, but numbers don't lie.
When we represent that energy storage is "green," that is batteries, or worse, hydrogen, what we are really talking about is destroying the exergy of dangerous fossil fuels, making the impact worse, not better.
The belief in these tiresome fantasies, that batteries are "green" or even worse, that hydrogen is "green" plays into the fact that degradation of the planetary atmosphere has reached the fastest pace ever observed in 2024, not that we really give a shit, since we're all promoting dangerous fossil fuels either to others (people rebranding fossil fuels as "hydrogen" and running sleazy ads) and/or batteries.
At the Mauna Loa CO2 Observatory, the 2024 Terror Continues.
Things are getting worse faster than ever.
Let me repeat: People lie to themselves and they lie to each other, but numbers don't lie.
The paper to which I'll refer in this post, is this one: Cleaning up while Changing Gears: The Role of Battery Design, Fossil Fuel Power Plants, and Vehicle Policy for Reducing Emissions in the Transition to Electric Vehicles Matthew Bruchon, Zihao Lance Chen, and Jeremy Michalek Environmental Science & Technology 2024 58 (8), 3787-3799
It's open sourced - anyone can read it - and refers to the grid on which I live, and in fact to the type of car I drive, an HEV, a hybrid Toyota Camry which routinely gets well over 50 mpg, sometimes (depending on the weather) close to or even exceeding 60 mpg, which doesn't make it "green," although it slightly ameliorates the filth associated with my lifestyle.
Again, it's open, and I'm not going to bother talking about but I can't help pointing to the pictures of the source of electricity on my grid (PJM) and other grids around the world, despite the very ignorant lie that electricity is "green."
The grid on which I live, by the way, the authors claim is "typical" although as the next graphic shows that there are definitely places that are even worse.

The caption:
You see the red regions in the graph. This is low carbon and reliable electricity, not dependent on the weather at a time the weather has been vastly destabilized by the unrestricted brown and black regions promoted by antinukes. And let's be clear again, the "bait and switch" game is what is promoted by people claiming that hydrogen is "green" and batteries are "green." What they're selling is fossil fuels.
A Giant Climate Lie: When they're selling hydrogen, what they're really selling is fossil fuels.
By the way, the investment in allegedly "green" so called "renewable energy" and the mining dependent distribution network designed to make what doesn't work, work although it's marked with decades of failure, easily outstrips the money being spent on the larger and reliable low carbon source of energy in red, which people claim is "too expensive" while ignoring the question of whether climate change is "too expensive."
The graphics in the following link are interactive, showing the "investments" in all forms of energy: IEA World Investment in Power
The, um, results of these vast expenditures are also graphic, albeit in another way.
The numbers are here: 2023 World Energy Outlook published by the International Energy Agency (IEA), Table A.1a on Page 264.
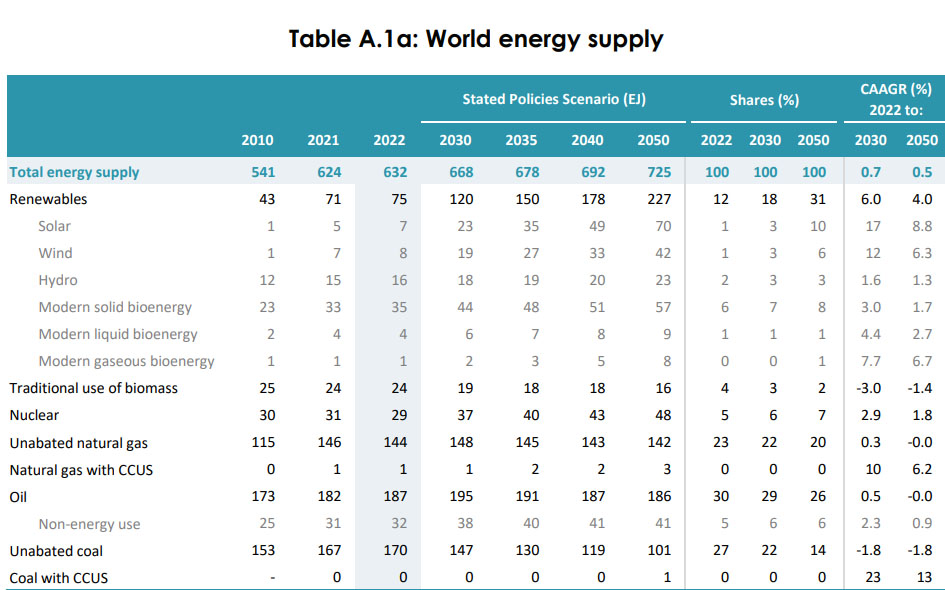
Trillions of dollars for 7 Exajoules of magical solar, and 8 Exajoules of magical wind power, all of which will be landfill in 20 to 25 years, if not sooner.
At long last, no sense of decency.
Anyway, about the external costs, the costs to the environment, human health, and ecosystem sustainability of cars:
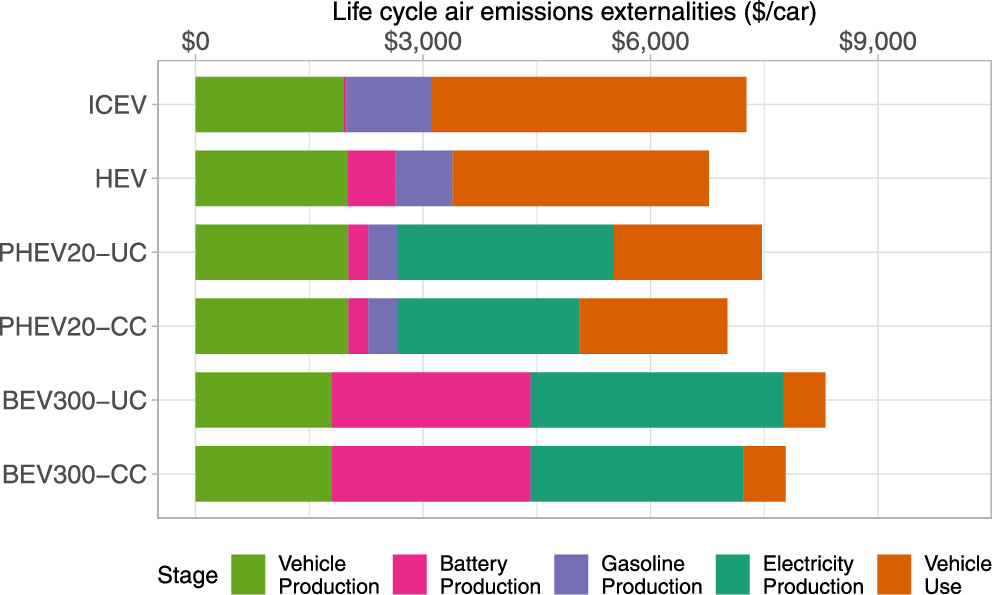
It looks like on my grid, my type of car is the least obnoxious, although all cars are obnoxious, including mine.
On and it's not like people really wait until the sun is shining and the wind is blowing to charge their "green" electric cars, is it?
I really shouldn't bother, should I? It's not like all this bitterness and anger at the end of my life will cause people to stop lying to themselves and lying to each other, selling batteries with ores dug by slaves, some of whom are children, or rebranding fossil fuels as "hydrogen," in order to waste energy and drive fossil fuel sales. The bull keeps flying around while things get worse.
Reality is unpopular.
We. Just. Don't. Give. A. Shit.
Trump may be the most famous egregious liar on the planet but he's hardly the only one.
Once again, people lie to themselves and they lie to each other, but numbers don't lie.
History will not forgive us, nor should it.
Enjoy the upcoming weekend.
Profile Information
Gender: MaleCurrent location: New Jersey
Member since: 2002
Number of posts: 33,516